RYTELO Powder for concentrate for solution for infusion Ref.[114877] Active ingredients: Imetelstat
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Geron Netherlands B.V., Naritaweg 165, 1043 BW Amsterdam, Netherlands
4.1. Therapeutic indications
Rytelo is indicated as monotherapy for the treatment of adult patients with transfusion-dependent anaemia due to very low, low or intermediate risk myelodysplastic syndromes (MDS) without an isolated deletion 5q cytogenetic (non-del 5q) abnormality and who had an unsatisfactory response to or are ineligible for erythropoietin-based therapy (see section 5.1).
4.2. Posology and method of administration
Rytelo should be administered and monitored under the supervision of physicians and healthcare professionals who are experienced in haematologic disease and treatment.
Complete blood cell count and liver function tests are recommended before administration of each dose. Additionally, weekly blood cell counts are recommended following the first two doses (see section 4.4).
A pregnancy test should be performed before administration of the first dose of Rytelo for females of reproductive potential (see section 4.6).
Posology
The recommended dose of Rytelo is 7.1 mg/kg body weight administered as an intravenous infusion once every 4 weeks. Rytelo should be discontinued if patients do not experience a reduction in red blood cell (RBC) transfusion burden after 24 weeks of treatment (6 doses) or if unacceptable toxicity occurs at any time.
Premedication for potential infusion-related reactions
Patients should be premedicated with diphenhydramine (25 to 50 mg) and hydrocortisone (100 to 200 mg), or equivalent, at least 30 minutes before dosing with Rytelo. Premedication should be administered before any doses of Rytelo, to prevent or reduce potential infusion-related reactions (see section 4.4).
Dose modifications
Recommended dose reductions for all Grade 3 and Grade 4 adverse reactions are found in Table 1.
The management of Grade 3 and Grade 4 adverse reactions may require a dose delay, dose reduction, or treatment discontinuation and are presented in Table 2, Table 3 and Table 4. Treatment with Rytelo should be permanently discontinued if the patient cannot tolerate the lowest dose level of 4.4 mg/kg.
Table 1. Recommended dose reduction for all Grade 3 and Grade 4 adverse reactions:
| Dose reduction | Current dose | Decreased dose |
|---|---|---|
| First dose reduction | 7.1 mg/kg | 5.6 mg/kg |
| Second dose reduction | 5.6 mg/kg | 4.4 mg/kg |
Grade 3 and Grade 4 haematologic adverse reactions
Delay administration of Rytelo if absolute neutrophil count is less than 1.0 × 109/L or platelets are less than 50 × 109/L. Modify dose as described in Table 2.
Table 2. Dose modifications for Grade 3 and Grade 4 haematologic adverse reactions:
| Adverse reaction | Severity gradea,b | Occurrence | Treatment modification |
|---|---|---|---|
| Thrombocytopenia (see sections 4.4 and 4.8) | Grade 3 | First | • Delay treatment until platelets are ≥ 50 × 109/L • Restart at same dose level |
| Second and third | • Delay treatment until platelets are ≥ 50 × 109/L • Restart at one dose level lower | ||
| Grade 4 | First and second | • Delay treatment until platelets are ≥ 50 × 109/L • Restart at one dose level lower | |
| Neutropenia (see sections 4.4 and 4.8) | Grade 3 | First | • Delay treatment until absolute neutrophil counts are ≥ 1.0 × 109/L • Restart at same dose level |
| Second and third | • Delay treatment until absolute neutrophil counts are ≥ 1.0 × 109/L • Restart at one dose level lower | ||
| Grade 4 | First and second | • Delay treatment until absolute neutrophil counts are ≥ 1.0 × 109/L • Restart at one dose level lower |
a Severity based on National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 4.03.
b Grade 3: severe; Grade 4: life-threatening
Non-haematologic adverse reactions
Dose administration modifications for infusion-related reactions are described in Table 3.
Table 3. Dose administration modifications for infusion-related reactions:
| Adverse reaction | Severity gradea,b | Occurrence | Treatment modification |
|---|---|---|---|
| Infusion-related reactions (see sections 4.4 and 4.8) | Grade 2 or 3 | First and second | • Interrupt the infusion until resolution or the intensity of the adverse reactions decrease to Grade 1b • Restart infusion at 50% of the infusion rate administered prior to the adverse reactions (i.e., 125 mL/h) |
| Third | • For Grade 2, stop infusion. May restart at next dose administration • For Grade 3, discontinue treatment | ||
| Grade 4 | First | • Stop infusion • Administer supportive care as appropriate and discontinue treatment |
a Severity based on NCI CTCAE, version 4.03.
b Grade 1: mild; Grade 2: moderate; Grade 3: severe; Grade 4: life-threatening
Dose modifications for other adverse drug reactions, including elevated liver function tests, are described in Table 4.
Table 4. Dose modifications for non-haematologic adverse reactions:
| Adverse reaction | Severity gradea,b | Occurrence | Treatment modification |
|---|---|---|---|
| Other adverse drug reactions including elevated liver function tests (see section 4.8) | Grade 3 or 4 | First and second | • Delay treatment until adverse reactions are Grade 1b or at baseline Grade • Restart at one dose level lower |
a Severity based on NCI CTCAE, version 4.03.
b Grade 1: mild; Grade 3: severe; Grade 4: life-threatening
Missed doses
If a planned dose is missed, the patient should be administered Rytelo as soon as possible and dosing continued as prescribed with 4 weeks between doses.
Special populations
Elderly
No dose adjustments are required in elderly patients (≥65 years of age).
Renal impairment
No dose adjustment is required in patients with mild to moderate renal impairment (creatinine clearance [CrCL] 30 to ˂90 mL/min). There is insufficient data in patients with severe renal impairment (CrCL 15 to ˂30 mL/min) or end-stage renal disease to support a dose recommendation (see section 5.2).
Hepatic impairment
No dose adjustment is required in patients with mildly to moderately abnormal liver function tests (total bilirubin ≤ upper limit of normal [ULN] and aspartate aminotransferase [AST] ˃ ULN or total bilirubin ˃ 1× to 1.5× ULN (Grade 1) and any AST) or (total bilirubin ˃ 1.5× to 3× ULN (Grade 2) and any AST). There is insufficient data in patients with severely abnormal liver function tests (total bilirubin ˃ 3× ULN (Grade 3) and any AST) to support a dose recommendation (see section 5.2).
Paediatric population
The safety and efficacy of Rytelo in children and adolescents aged 28 days to less than 18 years have not yet been established. No data are available.
There is no relevant use of Rytelo in paediatric patients aged less than 28 days.
Method of administration
Rytelo is for intravenous use.
Rytelo is provided for single use only.
4.9. Overdose
In case of overdose or incorrect administration (such as intravenous push or bolus), patients should be monitored for any signs or symptoms of adverse reactions, and appropriate symptomatic treatment and standard supportive care should be given.
6.3. Shelf life
Unopened vial
4 years.
Prepared solution
Reconstituted solution:
Use immediately to prepare the diluted solution for intravenous infusion. Diluted solution Use within 48 hours when stored refrigerated at 2°C to 8°C (includes the total time from the time of reconstitution to completion of the infusion).
Use within 18 hours when stored at room temperature at 20°C to 25°C (includes the total time from the time of reconstitution to completion of the infusion).
Chemical and physical in-use stability has been demonstrated for 48 hours at 2°C to 8°C or for 18 hours at 20°C to 25°C. From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 hours at 2°C to 8°C, unless reconstitution and dilution has taken place in controlled and validated aseptic conditions.
6.4. Special precautions for storage
Store in a refrigerator (2⁰C to 8⁰C).
Do not freeze.
For storage conditions after reconstitution and dilution of the medicinal product (see section 6.3).
6.5. Nature and contents of container
Rytelo 47 mg powder for concentrate for solution for infusion is a clear, 8 mL Type 1 glass vial with a chlorobutyl rubber stopper and an aluminium flip-off seal with a dark green plastic cap.
Pack size: 1 vial.
Rytelo 188 mg powder for concentrate for solution for infusion is a clear, 10 mL Type 1 glass vial with a chlorobutyl rubber stopper and an aluminium flip-off seal with a royal blue plastic cap.
Pack size: 1 vial.
6.6. Special precautions for disposal and other handling
For single use only.
Preparation, administration and handling instructions
Rytelo is provided as a white to off-white or slightly yellow lyophilised powder for intravenous infusion only and must be reconstituted and diluted prior to administration.
Reconstitution:
- Calculate the dose of Rytelo (total mg) based on the patient’s body weight (kg): Body Weight (X kg) × Dose (7.1 mg/kg, unless dose reduction to 5.6 mg/kg or 4.4 mg/kg is warranted).
- Determine the number of Rytelo vials needed. More than one vial may be needed to achieve a full dose. Each Rytelo vial contains 47 mg or 188 mg.
- Remove the Rytelo vials from the refrigerator. Allow the vials to sit for 10 to 15 minutes (not to exceed 30 minutes) to warm to room temperature 20°C to 25°C prior to reconstitution.
- Reconstitute each vial of Rytelo according to the strength and instructions specified below:
- Rytelo vials containing 47 mg powder for concentrate for solution for infusionη
Inject 1.8 mL of sodium chloride 9 mg/mL (0.9%) solution for injection directly onto the lyophilised powder for a deliverable volume of 1.5 mL. The final concentration of reconstituted solution will be 31.4 mg/mL per vial. - Rytelo vials containing 188 mg powder for concentrate for solution for infusionη
Inject 6.3 mL of sodium chloride 9 mg/mL (0.9%) solution for injection directly onto the lyophilised powder for a deliverable volume of 6.0 mL. The final concentration of reconstituted solution will be 31.4 mg/mL per vial.
- Rytelo vials containing 47 mg powder for concentrate for solution for infusionη
Each vial contains an overfill to account for loss of liquid during preparation and extraction of the reconstituted solution, resulting in the final concentration of 31.4 mg/mL specified above.
- Swirl each vial gently to avoid foaming until the powder is fully reconstituted (not to exceed 15 minutes). Do not shake.
- Visually inspect the reconstituted solution for particulate matter and discolouration prior to dilution. The reconstituted solution in each vial should appear as a clear to slightly hazy solution, essentially-free of visible contaminants, particles and/or particulates. Do not use if discolouration or particulate matter is present.
- Use the reconstituted solution immediately to prepare the Rytelo diluted solution in the infusion bag (see section 6.3).
Dilution:
- Calculate the volume of reconstituted solution required for the patient based on the patient’s body weight.
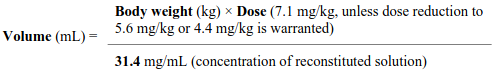
- To a 500 mL infusion bag of sodium chloride 9 mg/mL (0.9%) solution, add the required volume of reconstituted solution into the infusion bag. Discard any excess liquid remaining in the vial(s) which is not needed to achieve the required dose.
- Gently invert the infusion bag at least 5 times to ensure that the reconstituted solution is well-mixed. Do not shake the infusion bag prior to administration.
Storage of diluted solution:
- Use within 48 hours when stored refrigerated at 2°C to 8°C (includes the total time from the time of reconstitution to completion of the infusion), see section 6.3.
- Use within 18 hours when stored at room temperature at 20°C to 25°C (includes the total time from the time of reconstitution to completion of the infusion), see section 6.3.
Disposal:
- No special requirements for disposal. Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.