SKYRIZI 600 mg Concentrate for solution for infusion Ref.[113270] Active ingredients: Risankizumab
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: AbbVie Deutschland GmbH & Co. KG, Knollstrasse, 67061 Ludwigshafen, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, interleukin inhibitors
ATC code: L04AC18
Mechanism of action
Risankizumab is a humanised immunoglobulin G1 (IgG1) monoclonal antibody that selectively binds with high affinity to the p19 subunit of human interleukin 23 (IL-23) cytokine without binding to IL-12 and inhibits its interaction with the IL-23 receptor complex. IL-23 is a cytokine that is involved in inflammatory and immune responses. By blocking IL-23 from binding to its receptor, risankizumab inhibits IL-23-dependent cell signalling and release of proinflammatory cytokines.
Pharmacodynamic effects
In a study of subjects with psoriasis, expression of genes associated with the IL-23/IL-17 axis was decreased in the skin after single doses of risankizumab. Reductions in epidermal thickness, infiltration of inflammatory cells, and expression of psoriatic disease markers were also observed in psoriatic lesions.
In a Phase 2 study of subjects with Crohn’s disease, expression of genes associated with the IL-23/Th17 axis were decreased in gut tissue after multiple doses of risankizumab. Reductions in faecal calprotectin (FCP), serum C reactive protein (CRP) and IL-22 were also observed after multiple doses in Phase 3 induction studies in Crohn’s patients. Decreases in FCP, CRP and serum IL-22 were maintained out to week 52 of the maintenance study.
In a Phase 2b/3 study of subjects with ulcerative colitis, statistically significant and clinically meaningful reduction from baseline was observed in the inflammatory biomarkers, FCP and CRP, and in the IL-23 pathway-associated biomarker, serum IL-22, at week 12 of the induction study. Decreases in FCP, CRP and serum IL-22 were maintained out to week 52 of the maintenance study.
Clinical efficacy and safety
Crohn’s disease
The efficacy and safety of risankizumab were assessed in 1 419 subjects with moderately to severely active Crohn’s disease in three multicentre, randomised, double-blind, placebo-controlled clinical studies. Enrolled subjects were 16 years of age or older with a Crohn’s Disease Activity Index (CDAI) of 220 to 450, an average daily stool frequency (SF) ≥4 and/or average daily abdominal pain score (APS) ≥2, and a Simple Endoscopic Score for CD (SES-CD) of ≥6, or ≥4 for isolated ileal disease, excluding the narrowing component and confirmed by a central reviewer.
There were two 12-week intravenous induction studies (ADVANCE and MOTIVATE), which included a 12-week extension period for subjects who did not achieve SF/APS clinical response (≥30% decrease in SF and/or ≥30% decrease in APS and both not worse than baseline) at week 12. ADVANCE and MOTIVATE were followed by a 52-week randomised withdrawal study of subcutaneous maintenance treatment (FORTIFY) that enrolled subjects with SF/APS clinical response to intravenous induction treatment, representing at least 64 weeks of therapy.
ADVANCE and MOTIVATE
In studies ADVANCE and MOTIVATE, subjects were randomised to receive risankizumab at either 600 mg (recommended dose), 1 200 mg, or placebo, at week 0, week 4, and week 8.
In ADVANCE, 58% (491/850) subjects had failed or were intolerant to treatment with one or more biologic therapies (prior biologic failure), and 42% (359/850) had failed or were intolerant to therapy with conventional therapies but not biologic therapies (without prior biologic failure). In ADVANCE, among the subjects without prior biologic failure, (87%) 314/359 were naïve to biologic therapy and the remaining 13% had received a biologic but never failed or demonstrated intolerance. All patients in MOTIVATE had prior biologic failure.
In both studies, a greater proportion of subjects treated with risankizumab achieved the co-primary endpoints of clinical remission at week 12 and endoscopic response at week 12 compared to placebo. Enhanced SF/APS clinical response and clinical remission were significant as early as week 4 in subjects treated with risankizumab and continued to improve through week 12 (Table 2).
Table 2. Efficacy results in ADVANCE and MOTIVATE:
| ADVANCE | MOTIVATE | |||||
|---|---|---|---|---|---|---|
| Placebo intravenously (N=175) % | Risankizumab 600 mg intravenously (N=336) % | Treatment differenced (95% CI) | Placebo intravenously (N=187) % | Risankizumab 600 mg intravenously (N=191) % | Treatment differenced (95% CI) | |
| Co-primary endpoints | ||||||
| Clinical remission at week 12e | 22% | 43% | 22% [14%, 30%]a | 19% | 35% | 15% [6%, 24%]b |
| Endoscopic response at week 12f | 12% | 40% | 28% [21%, 35%]a | 11% | 29% | 18% [10%, 25%]a |
| Additional endpoints | ||||||
| Enhanced SF/APS clinical response at week 4g | 31% | 46% | 15% [6%, 23%]b | 32% | 45% | 14% [4%, 23%]c |
| Enhanced SF/APS clinical response at week 12g | 42% | 63% | 21% [12%, 30%]a | 39% | 62% | 23% [13%, 33%]a |
| CDAI <150 at week 4 | 10% | 18% | 8% [1%, 14%]c | 11% | 21% | 10% [2%, 17%]c |
| CDAI <150 at week 12 | 25% | 45% | 21% [12%, 29%]a | 20% | 42% | 22% [13%, 31%]a |
| Mucosal healing at week 12h | (N=173) 8% | (N=336) 21% | 14% [8%, 19%]a | (N=186) 4% | (N=190) 14% | 9% [4%, 15%]b |
| Endoscopic remission at week 12i | 9% | 24% | 15% [9%, 21%]a | 4% | 19% | 15% [9%, 21%]a |
a Statistically significant under multiplicity-control for risankizumab vs placebo comparison (p<0.001).
b Statistically significant under multiplicity-control for risankizumab vs placebo comparison (p≤0.01).
c Nominal p≤0.05 risankizumab vs placebo comparison.
d Adjusted treatment difference.
e Clinical remission based on SF/APS: average daily SF ≤2.8 and not worse than baseline and average daily AP score ≤1 and not worse than baseline.
f Endoscopic response: greater than 50% decrease in SES-CD from baseline, or a decrease of at least 2 points for subjects with a baseline score of 4 and isolated ileal disease.
g Enhanced SF/APS clinical response: ≥60% decrease in average daily SF and/or ≥35% decrease in average daily AP score and both not worse than Baseline, and/or clinical remission.
h Mucosal healing: SES-CD ulcerated surface subscore of 0 in subjects with a subscore of ≥1 at Baseline.
i Endoscopic remission: SES-CD ≤4 and at least a 2 point reduction versus Baseline and no subscore greater than 1 in any individual variable.
At week 12, a higher proportion of subjects treated with risankizumab achieved a decrease of at least 100 points in baseline CDAI compared to placebo (ADVANCE, risankizumab=60%, placebo=37%, p<0.001; MOTIVATE, risankizumab=60%, placebo=30%, p<0.001).
At week 12, a higher proportion of subjects treated with risankizumab achieved both enhanced SF/APS clinical response and endoscopic response at week 12 compared to placebo (ADVANCE, risankizumab=31%, placebo=8%, p<0.001; MOTIVATE, risankizumab=21%, placebo=7%, p<0.001).
The results for the co-primary endpoints for the subgroups (without allowing for multiplicity) of subjects with and without prior biologic failure are presented in Table 3.
Table 3. Efficacy results at week 12 in subgroups of subjects with prior biologic treatment failure and subjects without prior biologic failure in ADVANCE:
| ADVANCE | |||
|---|---|---|---|
| Placebo intravenously | Risankizumab 600 mg | Treatment difference (95% CI) | |
| Clinical remission per SF/AP Score | |||
| Prior biologic failure | 23% (N=97) | 41% (N=195) | 18% [7%, 29%] |
| Without prior biologic failure | 21% (N=78) | 48% (N=141) | 27% [15%, 39%] |
| Endoscopic response | |||
| Prior biologic failure | 11% (N=97) | 33% (N=195) | 21% [12%, 31%] |
| Without prior biologic failure | 13% (N=78) | 50% (N=141) | 38% [27%, 49%] |
CD-related hospitalisations:
Rates of CD-related hospitalisations through week 12 were lower in subjects treated with risankizumab compared to placebo (ADVANCE, risankizumab=3%, placebo=12%, p<0.001; MOTIVATE, risankizumab=3%, placebo=11%, p≤0.01).
FORTIFY
The maintenance study FORTIFY evaluated 462 subjects with SF/APS clinical response to 12 weeks of risankizumab intravenous induction treatment in studies ADVANCE and MOTIVATE. Subjects were randomised to continue to receive a maintenance regimen of risankizumab 360 mg subcutaneously (recommended dose), or risankizumab 180 mg subcutaneously every 8 weeks, or to withdraw from risankizumab induction and receive placebo subcutaneously every 8 weeks for up to 52 weeks.
The co-primary endpoints were clinical remission at week 52 and, endoscopic response at week 52. Co-primary endpoints were also measured in subjects with and without prior biologic failure (see Table 4).
Table 4. Efficacy results in FORTIFY at week 52 (64 weeks from initiation of induction dose):
| FORTIFY | |||
|---|---|---|---|
| Risankizumab intravenous induction/ Placebo subcutaneouslyf (N=164) % | Risankizumab intravenous induction/ Risankizumab 360 mg subcutaneously (N=141) % | Treatment difference (95% CI) | |
| Co-primary endpoints | |||
| Clinical remission | 40% | 52% | 15% [5%, 25%]a,g |
| Prior biologic failure | 34% (N=123) | 48% (N=102) | 14% [1%,27%] |
| Without prior biologic failure | 56% (N=41) | 62% (N=39) | 5% [-16%,27%] |
| Endoscopic response | 22% | 47% | 28% [19%, 37%]b,g |
| Prior biologic failure | 20% (N=123) | 44% (N=102) | 23% [11%, 35%] |
| Without prior biologic failure | 27% (N=41) | 54% (N=39) | 27% [6%, 48%] |
| Additional endpoints | |||
| Enhanced SF/APS clinical response | 49% | 59% | 13% [2%, 23%]e,g |
| Maintenance of clinical remissionh | (N=91) 51% | (N=72) 69% | 21% [6%, 35%]d,g |
| Endoscopic remission | 13% | 39% | 28% [20%, 37%]c,g |
| Mucosal healing | (N=162) 10% | (N=141) 31% | 22% [14%, 30%]c,g |
a Statistically significant under multiplicity-control for risankizumab vs placebo comparison (p≤0.01).
b Statistically significant under multiplicity-control for risankizumab vs placebo comparison (p<0.001).
c Nominal p<0.001 risankizumab vs placebo comparison without overall type I error control.
d Nominal p≤0.01 risankizumab vs placebo comparison without overall type I error control.
e Nominal p≤0.05 risankizumab vs placebo comparison without overall type I error control.
f The induction-only group consisted of subjects who achieved clinical response to risankizumab induction therapy and were randomised to receive placebo in the maintenance study (FORTIFY).
g Adjusted treatment difference.
h Maintenance of clinical remission: clinical remission at week 52 in subjects with clinical remission at week 0.
Deep remission (clinical remission and endoscopic remission) at week 52 was observed at higher rates in subjects treated with risankizumab intravenously/risankizumab subcutaneously compared to subjects who received risankizumab intravenously/placebo subcutaneously (28% vs. 10%, respectively, nominal p<0.001).
At week 52, a higher proportion of subjects treated with risankizumab intravenously/risankizumab subcutaneously achieved CDAI <150 compared to risankizumab intravenously/placebo subcutaneously (52% vs. 41%, respectively, nominal p≤0.01). A higher proportion of subjects treated with risankizumab intravenously/risankizumab subcutaneously achieved a decrease of at least 100 points in baseline CDAI score compared to subjects treated with risankizumab intravenously/placebo subcutaneously (62% vs. 48%, respectively, nominal p≤ 0.01).
91 subjects who did not demonstrate SF/APS clinical response 12 weeks after risankizumab induction in studies ADVANCE and MOTIVATE received subcutaneous 360 mg dose of risankizumab at week 12 and week 20. Of these subjects, 64% (58/91) achieved SF/APS clinical response at week 24; 33 of the subjects achieving SF/APS clinical response enrolled in FORTIFY and continued receiving risankizumab 360 mg subcutaneously every 8 weeks for up to 52 weeks. Among these subjects, 55% (18/33) achieved clinical remission and 45% (15/33) achieved endoscopic response at week 52.
During FORTIFY, 30 subjects had loss of response to risankizumab 360 mg subcutaneously treatment and received rescue treatment with risankizumab (1 200 mg intravenous single dose, followed by 360 mg subcutaneously every 8 weeks). Of these subjects, 57% (17/30) achieved SF/APS clinical response at week 52. In addition, 20% (6/30) and 34% (10/29) of subjects achieved clinical remission and endoscopic response at week 52, respectively.
Health-related and quality of life outcomes
Health-related quality of life was assessed by the Inflammatory Bowel Disease Questionnaire (IBDQ) and 36-Item Short Form Health Survey (SF-36). Improvement in fatigue was evaluated by the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-Fatigue) scale. Work productivity was assessed by the Work Productivity and Activity Impairment CD (WPAI-CD) Questionnaire.
At week 12 of ADVANCE and MOTIVATE, subjects treated with risankizumab achieved clinically meaningful improvements from baseline in IBDQ total score, all IBDQ domain scores (bowel symptoms, systemic function, emotional function, and social function), SF-36 Physical and Mental Component Summary Score, FACIT-Fatigue and WPAI-CD compared to placebo. For WPAI-CD greater reductions in impairment while working, overall work impairment, and activity impairment were demonstrated in ADVANCE; and greater reduction in activity impairment was demonstrated in MOTIVATE. These improvements were maintained in subjects treated with risankizumab intravenously/risankizumab subcutaneously in FORTIFY through week 52.
Ulcerative colitis
The efficacy and safety of risankizumab was assessed in subjects with moderately to severely active ulcerative colitis in two multicentre, randomised, double-blind, placebo-controlled clinical studies. Enrolled subjects were ≥18 and ≤80 years of age with adapted Mayo Score (aMS) of 5 to 9 (using the Mayo scoring system, excluding Physician’s Global Assessment) with an endoscopic subscore (ES) of 2 or 3 on screening endoscopy, confirmed by central review.
The 12-week intravenous induction study (INSPIRE) included a 12-week extension period for subjects who did not achieve clinical response [defined as a decrease from baseline in the aMS ≥2 points and ≥30% from baseline, and a decrease in rectal bleeding subscore (RBS) ≥1 or an absolute RBS ≤1] at Week 12. INSPIRE was followed by a 52-week randomised withdrawal study of subcutaneous maintenance treatment (COMMAND) that enrolled subjects with clinical response to 12 weeks of risankizumab intravenous induction treatment, representing at least 64 weeks of therapy.
INSPIRE
In study INSPIRE, 975 subjects were randomised and received either risankizumab 1 200 mg or placebo, at week 0, week 4, and week 8.
In INSPIRE, 52% (503/975) of subjects had failed (inadequate response or intolerance) one or more biologics therapies, JAK inhibitors, and/or S1P receptor modulators. Of these 503 subjects, 488 (97%) failed biologics and 90 (18%) failed JAK inhibitors.
Enrolled subjects were permitted to use a stable dose of oral corticosteroids (up to 20 mg/day prednisone or equivalent), immunomodulators, and aminosalicylates. At baseline in INSPIRE, 36% of subjects received corticosteroids, 17% of subjects received immunomodulators and 73% of subjects received aminosalicylates. Patient disease activity was moderate (aMS ≤7) in 58% of subjects and severe (aMS >7) in 42% of subjects.
In INSPIRE, a significantly greater proportion of subjects treated with risankizumab achieved the primary endpoint of clinical remission per aMS [defined as stool frequency subscore (SFS) ≤ 1, and not greater than baseline, RBS = 0, and ES ≤ 1 without evidence of friability] at week 12 compared to placebo (Table 5). Results of the primary endpoint and key secondary endpoints are listed in Table 5.
Table 5. Efficacy results in INSPIRE at week 12:
| Endpoint | Placebo intravenously (N=325) % | Risankizumab 1 200 mg intravenously (N=650) % | Treatment difference (95% CI) |
|---|---|---|---|
| Disease activity and UC symptoms | |||
| Clinical remissionab | 6% | 20% | 14%f [10%, 18%] |
| With biologic and/or JAK inhibitor failure | 4% (N=170) | 11% (N=333) | 7% [3%, 12%] |
| Without biologic and/or JAK inhibitor failure | 8% (N=155) | 30% (N=317) | 21% [15%, 28%] |
| Clinical responsec | 36% | 64% | 29%f [22%, 35%] |
| With biologic and/or JAK inhibitor failure | 31% (N=170) | 55% (N=333) | 24% [15%, 33%] |
| Without biologic and/or JAK inhibitor failure | 41% (N=155) | 74% (N=317) | 33% [24%, 42%] |
| Endoscopic and histologic assessment | |||
| Mucosal healingd | 12% | 37% | 24%f [19%, 29%] |
| With biologic and/or JAK inhibitor failure | 10% (N=170) | 26% (N=333) | 16% [9%, 22%] |
| Without biologic and/or JAK inhibitor failure | 14% (N=155) | 48% (N=317) | 33% [26%, 41%] |
| Histologic-endoscopic mucosal healinge | 8% | 24% | 17%f [12%, 21%] |
| With biologic and/or JAK inhibitor failure | 7% (N=170) | 16% (N=333) | 9% [3%, 14%] |
| Without biologic and/or JAK inhibitor failure | 8% (N=155) | 33% (N=317) | 25% [18%, 32%] |
a Primary endpoint
b Clinical remission per aMS: SFS ≤ 1, and not greater than baseline, RBS = 0, and ES ≤ 1 without evidence of friability
c Clinical response per aMS: decrease from Baseline ≥ 2 points and ≥ 30%, and a decrease in RBS ≥ 1 or an absolute RBS ≤ 1
d ES ≤ 1 without the evidence of friability
e ES ≤ 1 without the evidence of friability and Geboes score ≤ 3.1 (indicating neutrophil infiltration in <5% of crypts, no crypt destruction and no erosions, ulcerations or granulation tissue)
f p < 0.00001, adjusted treatment difference (95% CI)
Clinical disease activity and symptoms:
The partial adapted Mayo score (paMS) is composed of SFS and RBS. Clinical response per paMS is defined as a decrease of ≥1 point and ≥30% from Baseline and a decrease in RBS ≥1 or an absolute RBS ≤1. The results of clinical response per paMS over time in INSPIRE are shown in Figure 1. Onset of efficacy was rapid with a greater proportion of subjects treated with risankizumab achieving clinical response as early as week 4 compared to placebo (52% vs 31%, respectively, p<0.00001).
Figure 1. Proportion of subjects achieving clinical response per paMS over time in induction study INSPIRE:
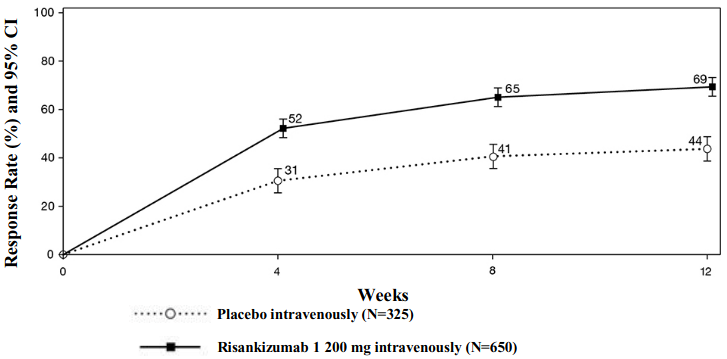
A significantly greater proportion of subjects treated with risankizumab compared to placebo had no abdominal pain (36% vs 26%, respectively, p<0.01) and no bowel urgency (44% vs 28%, respectively, p<0.00001) at week 12.
Other UC Symptoms:
Number of faecal incontinence episodes per week was reduced in a significantly greater amount in subjects treated with risankizumab compared to placebo at week 12 (change from baseline in risankizumab = -3.8, placebo = -2.2, p=0.00003).
The proportion of subjects who had no nocturnal bowel movements was significantly greater in subjects treated with risankizumab compared to placebo at week 12 (67% vs 43%, respectively, p<0.00001).
The proportion of subjects who had no tenesmus was significantly greater in subjects treated with risankizumab compared to placebo at week 12 (49% vs 30%, respectively, p<0.00001).
Number of days with sleep interruption due to UC symptoms per week were reduced in a significantly greater amount in subjects treated with risankizumab compared to placebo at week 12 (change from baseline in risankizumab = -2.5, placebo = -1.5, p<0.00001).
UC-related hospitalisations:
Rates of UC-related hospitalisations through week 12 were significantly lower in subjects treated with risankizumab compared to placebo (1% vs 6%, respectively, p<0.00001).
Extended treatment in week 12 non-responders:
A total of 141 subjects who did not demonstrate clinical response at week 12 of risankizumab induction in INSPIRE received either subcutaneous 180 mg or 360 mg dose of risankizumab at week 12 and week 20. Of the 71 subjects who received risankizumab 180 mg subcutaneously and 70 subjects who received risankizumab 360 mg subcutaneously, 56% and 57% achieved clinical response at week 24, respectively.
COMMAND
The maintenance study COMMAND evaluated 548 subjects with clinical response after 12 weeks of risankizumab intravenous induction treatment in study INSPIRE. Subjects were randomised to receive a maintenance regimen of risankizumab 180 mg subcutaneously or 360 mg subcutaneously every 8 weeks, or to withdraw from risankizumab induction and receive placebo subcutaneously every 8 weeks for up to 52 weeks.
In COMMAND, 75% (411/548) of subjects had failed (inadequate response or intolerance) one or more biologics therapies, JAK inhibitors, and/or S1P receptor modulators prior to induction baseline. Of these 411 subjects, 407 (99%) failed biologics and 78 (19%) failed JAK inhibitors.
In COMMAND, a significantly greater proportion of the above 548 subjects treated with risankizumab 180 mg subcutaneously or risankizumab 360 mg subcutaneously achieved the primary endpoint of clinical remission per aMS at week 52 compared to placebo (see Table 6). Results of the primary endpoint and key secondary endpoints are listed in Table 6.
Table 6. Efficacy results in COMMAND at week 52 (64 weeks from initiation of induction dose):
| Endpoint | Risankizumab intravenous induction/ Placebo subcutaneously+ (N=183) % | Risankizumab intravenous induction/ Risankizumab 180 mg subcutaneously (N=179) % | Risankizumab intravenous induction/ Risankizumab 360 mg subcutaneously (N=186) % | Treatment difference (97.5% CI)++ | ||
|---|---|---|---|---|---|---|
| Risankizumab intravenous induction/ Risankizumab 180 mg subcutaneously | Risankizumab intravenous induction/ Risankizumab 360 mg subcutaneously | |||||
| Disease activity and UC symptoms | ||||||
| Clinical remissionab | 25% | 40% | 38% | 16%h [6%, 27%] | 14%h [4%, 24%] | |
| With biologic and/or JAK inhibitor failure | 23% (N=138) | 37% (N=134) | 29% (N=139) | 13% [1%, 26%] | 6% [-6%, 18%] | |
| Without biologic and/or JAK inhibitor failure | 31% (N=45) | 51% (N=45) | 62% (N=47) | 20% [-3%, 43%] | 31% [8%, 53%] | |
| Maintenance of clinical remissionc | 40% (N=53) | 70% (N=44) | 50% (N=40) | 29%h [7%, 51%] | 13%k [-11%, 36%] | |
| With biologic and/or JAK inhibitor failure | 37% (N=35) | 65% (N=26) | 44% (N=25) | 28% [0%, 56%] | 7% [-22%, 36%] | |
| Without biologic and/or JAK inhibitor failure | 44% (N=18) | 77% (N=18) | 60% (N=15) | 33% [-2%, 67%] | 16% [-23%, 54%] | |
| Corticosteroid-free clinical remissiond | 25% | 40% | 37% | 16%h [6%, 26%] | 14%h [3%, 24%] | |
| With biologic and/or JAK inhibitor failure | 23% (N=138) 3 | 6% (N=134) | 29% (N=139) | 13% [0%, 25%] | 6% [-6%, 18%] | |
| Without biologic and/or JAK inhibitor failure | 31% (N=45) | 51% (N=45) | 60% (N=47) | 20% [-3%, 43%] | 28% [6%, 51%] | |
| Clinical responsee | 52% | 68% | 62% | 17%i [6%, 28%] | 11%j [0%, 23%] | |
| With biologic and/or JAK inhibitor failure | 46% (N=138) | 63% (N=134) | 57% (N=139) | 18% [4%, 31%] | 11% [-2%, 25%] | |
| Without biologic and/or JAK inhibitor failure | 71% (N=45) | 82% (N=45) | 79% (N=47) | 11% [-9%, 31%] | 8% [-13%, 28%] | |
| Endoscopic and histologic assessment | ||||||
| Mucosal healingf | 32% | 51% | 48% | 20%h [9%, 31%] | 17%h [7%, 28%] | |
| With biologic and/or JAK inhibitor failure | 30% (N=138) | 48% (N=134) | 39% (N=139) | 17% [4%, 30%] | 8% [-4%, 21%] | |
| Without biologic and/or JAK inhibitor failure | 36% (N=45) | 60% (N=45) | 76% (N=47) | 24% [1%, 47%] | 41% [19%, 62] | |
| Histologic-endoscopic mucosal healingg | 23% | 43% | 42% | 20%h [10%, 31%] | 20%h [10%, 30%] | |
| With biologic and/or JAK inhibitor failure | 22% (N=138) | 39% (N=134) | 33% (N=139) | 17% [5%, 29%] | 11% [-1%, 23%] | |
| Without biologic and/or JAK inhibitor failure | 29% (N=45) | 55% (N=45) | 69% (N=47) | 26% [3%, 49%] | 40% [19%, 62%] | |
+ The induction-only group consisted of subjects who achieved clinical response to risankizumab induction therapy and were randomised to receive placebo in the maintenance study (COMMAND).
++ Adjusted difference for the overall treatment difference.
a Primary endpoint
b Clinical remission per aMS: SFS ≤ 1, and not greater than baseline, RBS = 0, and ES ≤ 1 without evidence of friability
c Clinical remission per aMS at week 52 among subjects who achieved clinical remission at the end of induction treatment
d Clinical remission per aMS at week 52 and corticosteroid-free for ≥90 days
e Clinical response per aMS: decrease from Baseline ≥ 2 points and ≥ 30%, and a decrease in RBS ≥ 1 or an absolute RBS ≤ 1
f ES of ≤ 1 without the evidence of friability
g ES ≤ 1 without the evidence of friability and Geboes score ≤ 3.1 (indicating neutrophil infiltration in <5% of crypts, no crypt destruction and no erosions, ulcerations or granulation tissue)
h Statistically significant under multiplicity-control for risankizumab vs placebo comparison (p ≤ 0.01).
i Nominal p ≤ 0.01 risankizumab vs placebo comparison
j Nominal p ≤ 0.05 risankizumab vs placebo comparison
k p = 0.2234
Clinical disease activity and symptoms
A significantly greater proportion of subjects treated with risankizumab intravenously/risankizumab 180 mg subcutaneously compared to risankizumab intravenously/placebo had no abdominal pain (47% vs 30%, respectively, p<0.001) and no bowel urgency (54% vs 31%, respectively, p<0.00001) at week 52. A greater proportion of subjects treated with risankizumab intravenously/risankizumab 360 mg subcutaneously compared to risankizumab intravenously/placebo had no bowel urgency (49% vs 31%, respectively, p<0.001) at week 52, and a numerically higher proportion of subjects had no abdominal pain compared to risankizumab intravenously/placebo (38% vs 30%, respectively, p=0.0895) at week 52.
Other UC symptoms
The proportion of subjects who had no nocturnal bowel movements was greater in subjects treated with risankizumab intravenously/risankizumab 180 mg subcutaneously and risankizumab intravenously/risankizumab 360 mg subcutaneously compared to risankizumab intravenously/placebo at week 52 (42% and 43% vs 30%, p<0.01 and p<0.001, respectively).
The proportion of subjects who had no tenesmus was greater in subjects treated with risankizumab intravenously/risankizumab 180 mg subcutaneously and risankizumab intravenously/risankizumab 360 mg subcutaneously compared to risankizumab intravenously/placebo at week 52 (37% and 37% vs 23%, respectively, p<0.01).
UC-related hospitalisations
Occurrence of UC-related hospitalisations through week 52 were numerically lower in subjects treated with risankizumab intravenously/risankizumab 180 mg subcutaneously and risankizumab intravenously/risankizumab 360 mg subcutaneously compared to risankizumab intravenously/placebo (0.6 per 100 subject-years and 1.2 per 100 subject-years vs 3.1 per 100 subject-years, p=0.0949 and p=0.2531, respectively).
Endoscopic and histologic assessment
Endoscopic remission (normalisation of the endoscopic appearance of the mucosa) was defined as ES of 0. At week 12 of INSPIRE, a significantly greater proportion of subjects treated with risankizumab compared to placebo achieved endoscopic remission (11% vs 3%, respectively, p<0.00001). At week 52 of COMMAND, a significantly greater proportion of subjects treated with risankizumab intravenously/risankizumab 180 mg subcutaneously and risankizumab intravenously/risankizumab 360 mg subcutaneously compared to risankizumab intravenously/placebo achieved endoscopic remission (23% and 24% vs 15%, respectively, p<0.05).
Deep mucosal healing was defined as ES of 0 and Geboes score < 2.0 (indicating no neutrophil in crypts or lamina propria and no increase in eosinophil, no crypt destruction, and no erosions, ulcerations or granulation tissue). At week 12 of INSPIRE, a significantly greater proportion of subjects treated with risankizumab compared to placebo achieved deep mucosal healing (6% vs 1%, respectively, p<0.00001). At week 52 of COMMAND, a numerically higher proportion of subjects treated with risankizumab intravenously/risankizumab 180 mg subcutaneously and risankizumab intravenously/risankizumab 360 mg subcutaneously compared to risankizumab intravenously/placebo achieved deep mucosal healing (13% and 16% vs 10%, p=0.2062 and p=0.0618, respectively).
In COMMAND, maintenance of mucosal healing at week 52 (ES ≤1 without friability) was seen in a greater proportion of subjects treated with risankizumab intravenously/risankizumab 180 mg subcutaneously and risankizumab intravenously/risankizumab 360 mg subcutaneously compared to risankizumab intravenously/placebo among subjects who achieved mucosal healing at the end of induction (74% and 54% vs 47%, p<0.01 and p=0.5629, respectively).
Rescue treatment
During COMMAND, subjects who had loss of response to risankizumab subcutaneously treatment received rescue treatment with risankizumab (a single intravenous induction dose, followed by 360 mg subcutaneously every 8 weeks). Among these subjects, in the risankizumab 180 mg subcutaneous and risankizumab 360 mg subcutaneous treatment group, 85% (17/20) and 74% (26/35) achieved clinical response at week 52, respectively. In addition, 24% (6/25) and 35% (13/37) of subjects achieved clinical remission per aMS, and 38% (10/26) and 45% (17/38) of subjects achieved endoscopic improvement at week 52 in the risankizumab 180 mg subcutaneous and risankizumab 360 mg subcutaneous treatment group, respectively.
Week 24 responders
A total of 100 subjects did not demonstrate clinical response after 12 weeks of induction treatment, received either subcutaneous 180 mg (N=56) or 360 mg (N=44) dose of risankizumab at week 12 and week 20, demonstrated clinical response at week 24, and continued receiving risankizumab 180 mg or 360 mg subcutaneously every 8 weeks for up to 52 weeks in COMMAND. Among these subjects, 46% and 45% achieved clinical response per aMS at week 52, and 18% and 23% achieved clinical remission per aMS at week 52, for risankizumab 180 mg and 360 mg subcutaneously respectively.
Health-related and quality of life outcomes
Subjects treated with risankizumab achieved clinically meaningful improvements from baseline in the Inflammatory Bowel Disease Questionnaire (IBDQ) (bowel symptoms, systemic function, emotional function, and social function) compared to placebo. Changes from baseline in IBDQ total score at week 12 with risankizumab compared to placebo were 42.6 and 24.3, respectively. Changes from baseline in IBDQ total score at week 52 were 52.6, 50.3 and 35.0 in subjects treated with risankizumab intravenously/risankizumab 180 mg subcutaneously, risankizumab intravenously/risankizumab 360 mg subcutaneously and risankizumab intravenously/placebo, respectively.
Subjects receiving risankizumab experienced significantly greater improvement from baseline in fatigue, as measured by FACIT-F score at week 12 compared to placebo. Changes from baseline in FACIT-F score at Week 12 with risankizumab compared to placebo were 7.9 and 3.3, respectively. Changes from baseline in FACIT-F score at week 52 were 10.9, 10.3 and 7.0 in subjects treated with risankizumab intravenously/risankizumab 180 mg subcutaneously, risankizumab intravenously/risankizumab 360 mg subcutaneously and risankizumab intravenously/placebo, respectively.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Skyrizi in one or more subsets of the paediatric population in the treatment of Crohn’s disease and ulcerative colitis (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics of risankizumab was similar between plaque psoriasis and psoriatic arthritis, and between Crohn’s disease and ulcerative colitis.
Absorption
Risankizumab exhibited linear pharmacokinetics with dose-proportional increase in exposure across dose ranges of 18 to 360 mg and 0.25 to 1 mg/kg administered subcutaneously, and 200 to 1 800 mg and 0.01 to 5 mg/kg administered intravenously.
Following subcutaneous dosing of risankizumab, peak plasma concentrations were achieved between 3-14 days after dosing with an estimated absolute bioavailability of 74-89%. With dosing of 150 mg at week 0, week 4 and every 12 weeks thereafter, estimated steady-state peak and trough plasma concentrations are 12 and 2 μg/mL, respectively.
In subjects with Crohn’s disease treated with 600 mg intravenous induction dose at weeks 0, 4, and 8 followed by 360 mg subcutaneous maintenance dose at week 12 and every 8 weeks thereafter, maximum median peak and trough concentrations are estimated to be 156 and 38.8 μg/mL respectively during the induction period (weeks 8-12) and steady-state median peak and trough concentrations are estimated to be 28.0 and 8.13 μg/mL respectively during the maintenance period (weeks 40-48).
In subjects with ulcerative colitis treated with 1 200 mg intravenous induction dose at weeks 0, 4, and 8 followed by 180 mg or 360 mg subcutaneous maintenance dose at week 12 and every 8 week thereafter, maximum median peak and trough concentrations are estimated to be 350 and 87.7 μg/mL respectively during the induction period (weeks 8-12) and steady-state median peak and trough concentrations are estimated to be 19.6 and 4.64 μg/mL for the 180 mg subcutaneous dose and 39.2 and 9.29 μg/mL for the 360 mg subcutaneous dose, respectively, during the maintenance period (weeks 40-48).
Distribution
The mean (±standard deviation) steady-state volume of distribution (Vss) of risankizumab was 11.4 (±2.7) L in Phase 3 studies in subjects with psoriasis, indicating that the distribution of risankizumab is primarily confined to the vascular and interstitial spaces. In a typical 70 kg subject with Crohn’s disease, Vss was 7.68 L.
Biotransformation
Therapeutic IgG monoclonal antibodies are typically degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgGs. Risankizumab is not expected to be metabolised by cytochrome P450 enzymes.
Elimination
The mean (±standard deviation) systemic clearance (CL) of risankizumab was 0.3 (±0.1) L/day in Phase 3 studies in subjects with psoriasis. The mean terminal elimination half-life of risankizumab ranged from 28 to 29 days in Phase 3 studies in subjects with psoriasis. For a typical 70 kg subject with Crohn’s disease, CL was 0.30 L/day and terminal elimination half-life was 21 days.
As an IgG1 monoclonal antibody, risankizumab is not expected to be filtered by glomerular filtration in the kidneys or to be excreted as an intact molecule in the urine.
Linearity/non-linearity
Risankizumab exhibited linear pharmacokinetics with approximately dose-proportional increases in systemic exposure (Cmax and AUC) in the evaluated dose ranges of 18 to 360 mg or 0.25 to 1 mg/kg subcutaneous administration and 200 to 1 800 mg and 0.01 to 5 mg/kg administered intravenously in healthy subjects or subjects with psoriasis, Crohn’s disease or ulcerative colitis.
Interactions
Interaction studies were conducted in subjects with plaque psoriasis, Crohn’s disease, or ulcerative colitis to assess the effect of repeated administration of risankizumab on the pharmacokinetics of cytochrome P450 (CYP) sensitive probe substrates. The exposure of caffeine (CYP1A2 substrate), warfarin (CYP2C9 substrate), omeprazole (CYP2C19 substrate), metoprolol (CYP2D6 substrate) and midazolam (CYP3A substrate) following risankizumab treatment were comparable to their exposures prior to risankizumab treatment, indicating no clinically meaningful interactions through these enzymes.
Population pharmacokinetic analyses indicated that risankizumab exposure was not impacted by concomitant medicinal products used by some subjects with plaque psoriasis during the clinical studies. Similar lack of impact by concomitant medicinal products was observed based on population pharmacokinetic analyses in Crohn’s disease or ulcerative colitis.
Special populations
Paediatric population
The pharmacokinetics of risankizumab in paediatric subjects under 16 years of age has not been established. Of the 1 574 subjects with Crohn’s disease exposed to risankizumab, 12 were 16 to 17 years old. Risankizumab exposures in 16 to 17 year-old subjects with Crohn’s disease were similar to those in adults. Age was not found to have any significant impact on risankizumab exposures based on the population pharmacokinetic analyses.
Elderly
Of the 2 234 subjects with plaque psoriasis exposed to risankizumab, 243 were 65 years or older and 24 subjects were 75 years or older. Of the 1 574 subjects with Crohn’s disease exposed to risankizumab, 72 were 65 years or older and 5 subjects were 75 years or older. Of the 1 512 subjects with ulcerative colitis exposed to risankizumab, 103 were 65 years or older and 8 subjects were 75 years or older. No overall differences in risankizumab exposure were observed between older and younger subjects who received risankizumab.
Patients with renal or hepatic impairment
No specific studies have been conducted to determine the effect of renal or hepatic impairment on the pharmacokinetics of risankizumab. Based on population pharmacokinetic analyses, serum creatinine levels, creatinine clearance, or hepatic function markers (ALT/AST/bilirubin) did not have a meaningful impact on risankizumab clearance in subjects with psoriasis, Crohn’s disease, or ulcerative colitis.
As an IgG1 monoclonal antibody, risankizumab is mainly eliminated via intracellular catabolism and is not expected to undergo metabolism via hepatic cytochrome P450 enzymes or renal elimination.
Body weight
Risankizumab clearance and volume of distribution increase as body weight increases which may result in reduced efficacy in subjects with high body weight (>130 kg). However, this observation is based on a limited number of subjects with plaque psoriasis. Body weight had no clinically meaningful impact on risankizumab exposure or efficacy in psoriatic arthritis, Crohn’s disease, or ulcerative colitis. No dose adjustment based on body weight is currently recommended.
Gender or race
The clearance of risankizumab was not significantly influenced by gender or race in adult subjects with plaque psoriasis, Crohn’s disease or ulcerative colitis. No clinically meaningful differences in risankizumab exposure were observed in Chinese or Japanese subjects compared to Caucasian subjects in clinical pharmacokinetic studies in healthy volunteers.
5.3. Preclinical safety data
Nonclinical data revealed no special hazard for humans based on repeat-dose toxicity studies including safety pharmacology evaluations and an enhanced pre- and post-natal developmental toxicity study in cynomolgus monkeys at doses of up to 50 mg/kg/week, producing exposures 10 times the clinical exposures during induction at a dose of 600 mg intravenous every 4 weeks and 39 times the clinical exposures for maintenance when given 360 mg subcutaneously every 8 weeks for Crohn’s disease. For ulcerative colitis, exposures were 5 times the clinical exposures during induction at a dose of 1 200 mg intravenously every 4 weeks and 65 or 32 times the clinical exposures for maintenance when given 180 or 360 mg subcutaneously every 8 weeks.
Mutagenicity and carcinogenicity studies have not been conducted with risankizumab. In a 26-week chronic toxicology study in cynomolgus monkeys at doses of up to 50 mg/kg/week (7 times the clinical exposures during induction at a dose of 600 mg intravenous every 4 weeks and 28 times the clinical exposures for maintenance when given 360 mg subcutaneously every 8 weeks for Crohn’s disease and 3 times the clinical exposures during induction at a dose of 1 200 mg intravenously every 4 weeks and 45 or 23 times the clinical exposures for maintenance when given 180 or 360 mg subcutaneously every 8 weeks for ulcerative colitis), there were no pre-neoplastic or neoplastic lesions observed and no adverse immunotoxicity or cardiovascular effects were noted.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.