SPEVIGO Concentrate for solution for infusion Ref.[50688] Active ingredients: Spesolimab
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Boehringer Ingelheim International GmbH, Binger Str. 173, 55216 Ingelheim am Rhein, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, interleukin inhibitors
ATC code: L04AC22
Mechanism of action
Spesolimab is a humanised antagonistic monoclonal immunoglobulin G1 (IgG1) antibody blocking human interleukin 36 receptor (IL36R) signalling. Binding of spesolimab to IL36R prevents the subsequent activation of IL36R by its ligands (IL36 α, β and γ) and downstream activation of pro-inflammatory pathways.
Pharmacodynamic effects
Following treatment with intravenous spesolimab in patients with GPP, reduced levels of C-reactive protein (CRP), IL6, T helper cell (Th1/Th17) mediated cytokines, keratinocyte-mediated inflammation markers, neutrophilic mediators, and proinflammatory cytokines were observed in serum and skin at week 1 compared to baseline and were associated with a decrease in clinical severity. These reductions in biomarkers became more pronounced at the last measurement at week 8 in Effisayil 1.
Clinical efficacy and safety
Effisayil 1 (1368-0013)
A randomised, double-blind, placebo-controlled study (Effisayil 1) was conducted to evaluate the clinical efficacy and safety of spesolimab in adult patients with flares of Generalised Pustular Psoriasis (GPP), as diagnosed per European Rare And Severe Psoriasis Expert Network (ERASPEN) criteria, regardless of IL36RN mutation status. Patients were randomised if they had a flare of GPP of moderate-to-severe intensity, as defined by a Generalised Pustular Psoriasis Physician Global Assessment (GPPGA) total score (which ranges from 0 [clear] to 4 [severe]) of at least 3 (moderate), presence of fresh pustules (new appearance or worsening of pustules), GPPGA pustulation sub score of at least 2 (mild), and at least 5% of body surface area covered with erythema and the presence of pustules. Patients were required to discontinue systemic and topical therapy for GPP prior to randomisation (see Table 2). Patients with an immediate life-threatening flare of GPP or requiring intensive care treatment were excluded from the study.
Table 2. Minimum time between discontinuation of restricted medications for GPP treatment and randomisation (Effisayil 1)*:
| Duration of washout period | Medications or class of medications |
|---|---|
| 2 months | adalimumab, alemtuzumab, briakinumab, brodalumab, efalizumab, guselkumab, infliximab, ixekizumab, natalizumab, risankizumab, rituximab, secukinumab, tildrakizumab, ustekinumab, visilizumab, investigational products for psoriasis (non biologics) |
| 6 weeks | etanercept |
| 30 days | systemic immunomodulatory treatments (e.g. corticosteroids**, cyclophosphamide), tofacitinib, apremilast; other systemic psoriasis treatments (e.g. fumarates), any investigational device or product (excluding psoriasis products); photochemotherapy (e.g. PUVA); granulocytes and monocytes adsorptive apharesis |
| 7 days | anakinra |
* No treatment initiation 1 week prior to randomisation: phototherapy (e.g. UVA, UVB), topical treatment for psoriasis or any other skin condition (e.g. topical corticosteroids, topical vitamin D analogues, tar, anthralin, topical retinoids); no treatment initiation 2 weeks prior to randomisation, no dose escalation within 2 weeks prior to randomisation, and had to be discontinued prior to receiving the first dose: methotrexate, cyclosporine, retinoids.
** No restriction on inhaled corticosteroids to treat asthma or corticosteroid drops administered in the eye or ear.
The primary endpoint of the study was the proportion of patients with a GPPGA pustulation sub score of 0 (indicating no visible pustules) at week 1 after treatment. The key secondary endpoint of the study was the proportion of patients with a GPPGA total score of 0 or 1 (clear or almost clear skin) at week 1. For the GPPGA pustulation sub score of 0 and the GPPGA total score of 0/1, non-responder imputation was used to handle the occurrence of escape (treatment at the investigator’s choice if the disease worsened) and rescue (single 900 mg dose of intravenous spesolimab) medication use and missing data.
A total of 53 patients were randomised (2:1) to receive a single intravenous dose of 900 mg spesolimab (n=35) or placebo (n=18). Patients in either treatment arm who still experienced flare symptoms at week 1 were eligible to receive a single intravenous dose of open-label 900 mg spesolimab, resulting in 12 patients (34%) in the spesolimab arm receiving a second dose of spesolimab and 15 patients (83%) in the placebo arm receiving one dose of spesolimab on day 8. In addition, 6 patients (4 spesolimab arm; 2 placebo arm) received flare treatment with a single 900 mg dose of intravenous spesolimab for reoccurrence of a flare after day 8.
The study population consisted of 32% men and 68% women. The mean age was 43 (range: 21 to 69) years; 55% of patients were Caucasian and 45% were Asian. Most patients included in the study had a GPPGA pustulation sub score of 3 (43%) or 4 (36%), and patients had a GPPGA total score of 3 (81%) or 4 (19%). 24.5% of patients had been previously treated with biologic therapy for GPP.
Primary and key secondary efficacy
At week 1, there was a statistically significant difference in the proportion of patients achieving a GPPGA pustulation sub score of 0 (indicating no visible pustules) and GPPGA total score of 0 or 1 (clear or almost clear skin) in the spesolimab arm compared with placebo (see Table 3).
Table 3. GPPGA pustulation sub score and GPPGA total score at week 1 (Effisayil 1):
| Placebo | Spesolimab 900 mg i.v. | |
|---|---|---|
| Number of Patients analysed | 18 | 35 |
| Patients achieving a GPPGA pustulation sub score of 0, n (%) | 1 (5.6) | 19 (54.3) |
| p-value* | 0.0004 | |
| Patients achieving a GPPGA total score of 0 or 1, n (%) | 2 (11.1) | 15 (42.9) |
| p-value* | 0.0118 | |
GPPGA = Generalised Pustular Psoriasis Physician Global Assessment; i.v. = intravenous
* One-sided p-value
For both the primary and the key secondary endpoint, treatment effect was observed for all patients regardless of the IL36RN mutation status.
Effisayil 2 (1368-0027)
A randomised, double-blind, placebo-controlled phase II b study (Effisayil 2) evaluated the efficacy and safety of spesolimab for subcutaneous administration in adult and adolescent patients with a history of GPP, as diagnosed per ERASPEN criteria, regardless of IL36RN mutation status, and with at least two GPP flares of moderate-to-severe intensity in the past. Patients were randomised if they had a GPPGA total score of 0 or 1 at screening and randomisation. Patients were required to discontinue systemic and topical therapy for GPP prior to or at randomisation. These patients must have had a history of flaring while on concomitant treatment for GPP or a history of flaring upon dose reduction or discontinuation of these concomitant medications.
The primary endpoint of the study was the time to the first GPP flare up to week 48 (defined by a GPPGA pustulation subscore of ≥2 and an increase in GPPGA total score by ≥2 from baseline). The key secondary endpoint of the study was the occurrence of at least one GPP flare up to week 48. Additional secondary endpoints at week 48 were the time to the first worsening of Psoriasis Symptom Scale (PSS) and Dermatology Quality of Life Index (DLQI) defined as a 4-point increase in total score from baseline.
A total of 123 patients were randomised (1:1:1:1) to receive one of the four treatments (see Table 4).
Table 4. Treatment arms in Effisayil 2:
| Loading dose | Subsequent doses | |
|---|---|---|
| spesolimab | 600 mg subcutaneously | 300 mg subcutaneously every 4 weeks |
| spesolimab | 600 mg subcutaneously | 300 mg subcutaneously every 12 weeks |
| spesolimab | 300 mg subcutaneously | 150 mg subcutaneously every 12 weeks |
| Placebo | subcutaneous treatment | subcutaneous treatment every 4 weeks |
The study population consisted of 38.2% men and 61.8% women. The mean age was 40.4 (range: 14 to 75) years with 8 (6.5%) adolescent patients (2 per treatment arm); 64.2% of patients were Asian and 35.8% were Caucasian. Patients included in the study had a GPPGA pustulation sub score of 1 (28.5%) or 0 (71.5%), and patients had a GPPGA total score of 1 (86.2%) or 0 (13.8%). At the time of randomisation, 74.8% of patients were treated with systemic therapy for GPP, which was discontinued at the start of the randomised study treatment.
While 3 dosing regimens were studied in Effisayil 2, the recommended dosing regimen for GPP flare prevention is a subcutaneous loading dose of 600 mg spesolimab followed by 300 mg subcutaneous treatment administered every 4 weeks (see section 4.2). The results summarised below are those for the recommended dosing regimen.
Patients who experienced a flare were eligible to receive up to two open-label, intravenous doses of 900 mg spesolimab (see section 4.2). 2 (6.7%) patients in the spesolimab arm for the recommended dose and 15 (48.4%) patients in the placebo arm received intravenous flare treatment.
Treatment with the recommended spesolimab dose compared to placebo resulted in statistically significant improvement based on the primary and key secondary endpoint (see Table 5).
Table 5. Time to the first GPP flare and occurrence of at least one GPP flare up to week 48 (Effisayil 2):
| Placebo | Recommended spesolimab dose | |
|---|---|---|
| Number of patients analysed, N | 31 | 30 |
| Patients with GPP flares, N (%)* | 16 (51.6) | 3 (10.0) |
| Hazard ratio (HR)** for the time to the first flare vs placebo (95% CI) | 0.16 (0.05, 0.54) | |
| p-value*** | 0.0005 | |
| Risk difference for GPP flare occurrence vs placebo (95% CI) | -39.0% (-62.1, -15.9) | |
| p-value**** | 0.0013 | |
* The use of intravenous spesolimab treatment or investigator-prescribed standard of care to treat GPP worsening were considered as onset of GPP flare
** Cox regression model stratified by the use of systemic GPP medications at randomisation
*** Log-rank test stratified by the use of systemic GPP medications at randomisation, one-sided p-value
**** Cochran-Mantel-Haenszel test after multiple imputation, stratified by the use of systemic GPP medications at randomisation, one-sided p-value
The efficacy of the subcutaneous recommended spesolimab dose compared with placebo was observed shortly after randomisation and was maintained up to week 48 (see Figure 1).
Figure 1. Time to the first GPP flare up to week 48 (Effisayil 2):
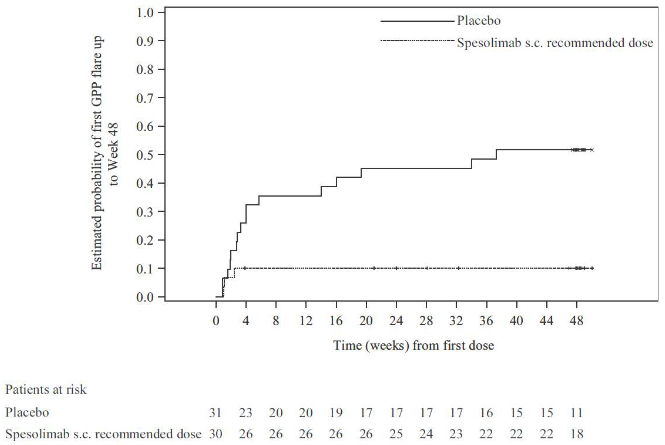
For both primary and key secondary endpoint, treatment effect was observed for all patients regardless of the IL36RN mutation status.
One adolescent patient in the placebo arm received investigator-prescribed standard of care to treat GPP worsening and was considered to have a GPP flare. No adolescent patient in the recommended spesolimab dose arm experienced a GPP flare.
The prevention of GPP worsening in terms of PSS, and DLQI was also observed, as shown by the hazard ratios for PSS 0.42 (95% CI 0.20, 0.91) and for DLQI 0.26 (95% CI 0.11, 0.62).
Immunogenicity
In patients with GPP treated with intravenous spesolimab in Effisayil 1, 46% of patients developed ADAs. A majority of ADA-positive subjects also developed neutralising antibodies. In Effisayil 2, following multiple subcutaneous doses of spesolimab, 41% of the patients developed ADAs. A majority of ADA-positive subjects also developed neutralising antibodies. Clearance of spesolimab increased along with increasing ADA titers.
As the majority of patients did not experience a subsequent new flare in Effisayil 1, the data on re-treatment of patients with ADA (n=4) is limited. It is currently unknown if there is a correlation between the presence of ADA to spesolimab and maintenance of efficacy for flare treatment. After subcutaneous administration of spesolimab in Effisayil 2, there was no apparent impact of ADA presence on efficacy or safety.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Spevigo in the paediatric population younger than 12 years of age in the treatment of generalised pustular psoriasis (see section 4.2 for information on paediatric use).
Conditional approval
This medicinal product has been authorised under a so-called ‘conditional approval’ scheme. This means that further evidence on this medicinal product is awaited. The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
A population pharmacokinetic model was developed based on data collected from healthy subjects, patients with GPP and patients with other diseases. After a single intravenous dose of 900 mg, the population PK model-estimated AUC0-∞ (95% CI) and Cmax (95% CI) in a typical ADA-negative patient with GPP were 4 750 (4 510, 4 970) μg·day/mL and 238 (218, 256) μg/mL, respectively. After a 600 mg subcutaneous loading dose of spesolimab followed by 300 mg spesolimab subcutaneously every 4 weeks, the mean (CV%) steady-state trough concentration ranged from 33.4 μg/mL (37.6%) to 42.3 μg/mL (43.0%).
Absorption
Following subcutaneous single dose administration of spesolimab in healthy volunteers, peak plasma concentrations were achieved between 5.5 to 7.0 days after dosing. After subcutaneous administration in the abdomen, absolute bioavailability was slightly higher at higher doses with estimated values of 58%, 65%, and 72% at 150 mg, 300 mg, and 600 mg, respectively. Based on limited data, absolute bioavailability in the thigh was approximately 85% following a subcutaneous dose of 300 mg spesolimab.
Distribution
Based on the population pharmacokinetic analysis, the typical volume of distribution at steady state was 6.4 L.
Biotransformation
The metabolic pathway of spesolimab has not been characterised. As a humanised IgG1 monoclonal antibody, spesolimab is expected to be degraded into small peptides and amino acids via catabolic pathways in a manner similar to endogenous IgG.
Elimination
In the linear dose range (0.3 to 20 mg/kg), based on the population PK model, spesolimab clearance (95% CI) in a typical ADA-negative patient with GPP, weighing 70 kg was 0.184 L/day. The terminal-half-life was 25.5 days.
Linearity/non-linearity
When administered intravenously, spesolimab exhibited linear pharmacokinetics with dose-proportional increase in exposure across single dose ranges of 0.3 to 20 mg/kg. Both clearance (CL) and terminal half-life were independent of dose. Following subcutaneous single dose administration, spesolimab exposure increased slightly more than dose-proportionally across the dose range of 150 mg to 600 mg due to slightly increased bioavailability at higher doses.
Body weight
Spesolimab concentrations were lower in subjects with higher body weight and higher in subjects with lower body weight. Spesolimab has not been studied in patients with GPP weighing more than 164 kg. Based on pharmacokinetic modelling and simulation, the recommended dose for adolescents from 12 years of age weighing ≥30 and <40 kg is half the recommended dose than for adults and adolescents from 12 years of age and weighing at least 40 kg (see section 4.2).
The exposure in patients weighing ≥30 and <40 kg receiving the reduced dosing regimen is expected to be comparable with those observed in GPP studies.
Elderly/gender/race
Based on population pharmacokinetic analyses, age, gender and race do not have a clinically relevant effect on the pharmacokinetics of spesolimab.
Hepatic and renal impairment
As a monoclonal antibody, spesolimab is not expected to undergo hepatic or renal elimination. No formal trial of the effect of hepatic or renal impairment on the pharmacokinetics of spesolimab was conducted.
Population PK analysis did not identify mild hepatic impairment or mild or moderate renal impairment as having an influence on the systemic exposure of spesolimab.
Paediatric population
The pharmacokinetics of spesolimab in paediatric patients below the age of 14 years have not been studied.
The plasma pharmacokinetics of spesolimab observed in adolescents were consistent with that observed in adults.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on repeated dose toxicity studies.
Developmental and reproductive toxicity
Non-clinical studies conducted in mice using a surrogate antibody directed towards murine IL36R do not indicate direct or indirect harmful effects with respect to pregnancy, embryonic/foetal development or fertility.
Genotoxicity
Genotoxicity studies have not been conducted with spesolimab.
Carcinogenicity
Carcinogenicity and mutagenicity studies have not been conducted with spesolimab.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.