SUNLENCA Solution for injection Ref.[50154] Active ingredients: Lenacapavir
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: Gilead Sciences Ireland UC, Carrigtohill, County Cork, T45 DP77, Ireland
4.3. Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
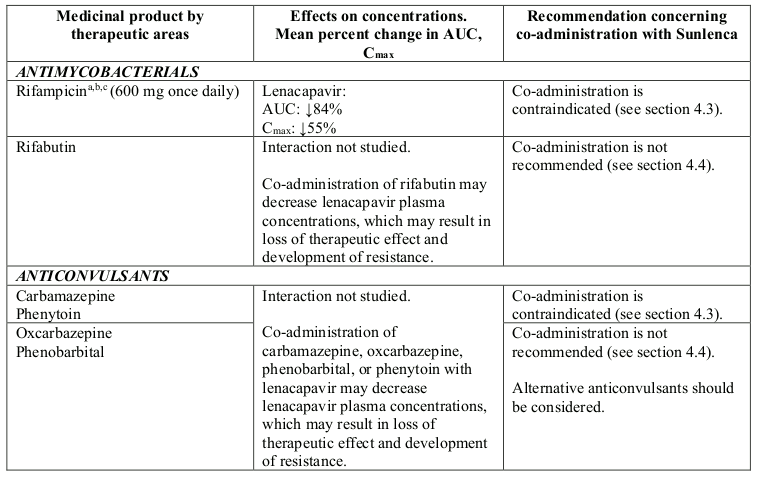
Co-administration with strong inducers of CYP3A, P-gp, and UGT1A1, such as:
- antimycobacterials: rifampicin
- anticonvulsants: carbamazepine, phenytoin
- herbal products: St. John’s wort (Hypericum perforatum)
(see section 4.5).
4.4. Special warnings and precautions for use
Risk of resistance following treatment discontinuation
If Sunlenca is discontinued, to minimise the risk of developing viral resistance it is essential to adopt an alternative, fully suppressive antiretroviral regimen where possible, no later than 28 weeks after the final injection of Sunlenca.
If virologic failure is suspected, an alternative regimen should be adopted where possible.
Use of other medicinal products after discontinuation of lenacapavir
If Sunlenca is discontinued, residual concentrations of lenacapavir may remain in the systemic circulation of patients for prolonged periods. These concentrations may affect the exposures of other medicinal products (i.e. sensitive CYP3A substrates) that are initiated within 9 months after the last subcutaneous dose of Sunlenca (see section 4.5). These concentrations are not expected to affect the exposures of other antiretroviral agents that are initiated after discontinuation of Sunlenca.
Immune Reconstitution Inflammatory Syndrome
In HIV infected patients with severe immune deficiency at the time of institution of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of CART. Relevant examples include cytomegalovirus retinitis, generalised and/or focal mycobacterial infections, and Pneumocystis jirovecii pneumonia. Any inflammatory symptoms should be evaluated and treatment instituted when necessary.
Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment.
Opportunistic infections
Patients should be advised that Sunlenca or any other antiretroviral therapy does not cure HIV infection and that they may still develop opportunistic infections and other complications of HIV infection. Therefore, patients should remain under close clinical observation by physicians experienced in the treatment of patients with HIV associated diseases.
Co-administration of other medicinal products
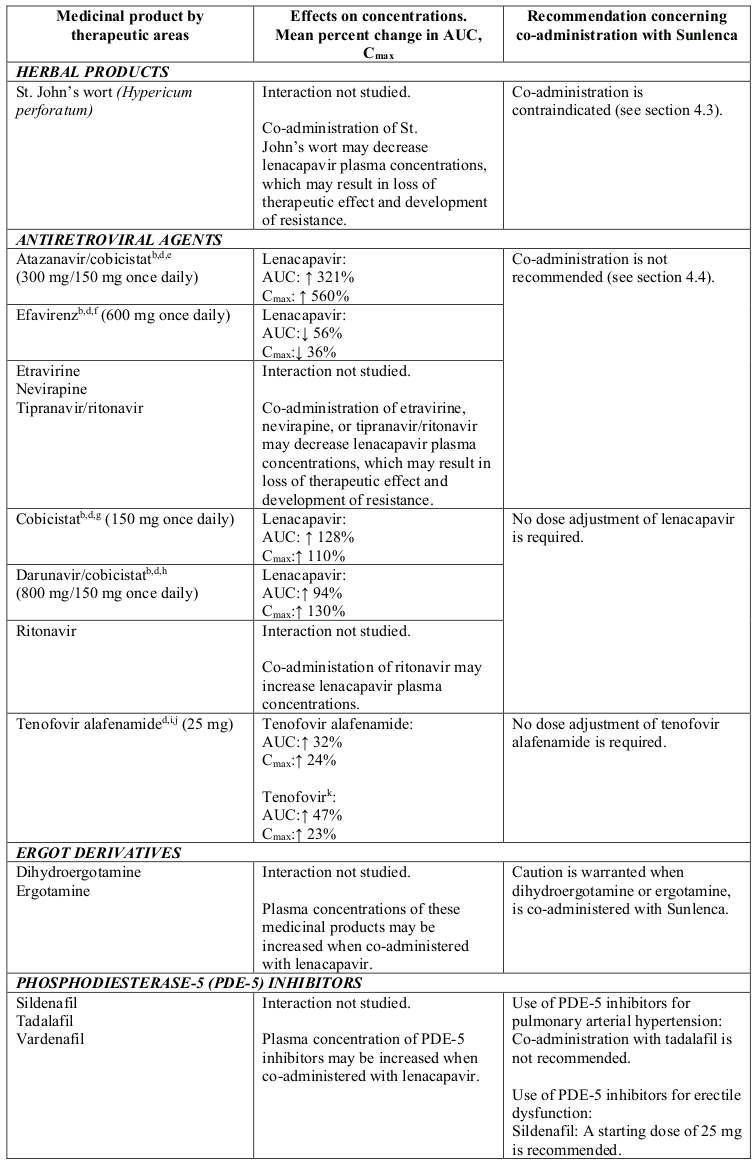
Co-administration with medicinal products that are moderate inducers of CYP3A and P-gp (e.g. efavirenz) is not recommended (see section 4.5).
Co-administration with medicinal products that are strong inhibitors of CYP3A, P-gp, and UGT1A1 together (i.e. all 3 pathways), such as atazanavir/cobicistat is not recommended (see section 4.5).
Excipients
This medicinal product contains less than 1 mmol sodium (23 mg) per injection, that is to say essentially ‘sodium-free’.
4.5. Interaction with other medicinal products and other forms of interaction
Effect of other medicinal products on the pharmacokinetics of lenacapavir
Lenacapavir is a substrate of CYP3A, P-gp and UGT1A1. Strong inducers of CYP3A, P-gp, and UGT1A1, such as rifampicin, may significantly decrease plasma concentrations of lenacapavir resulting in loss of therapeutic effect and development of resistance, therefore co-administration is contraindicated (see section 4.3). Moderate inducers of CYP3A and P-gp, such as efavirenz, may also significantly decrease plasma concentrations of lenacapavir, therefore co-administration is not recommended (see section 4.4).
Strong inhibitors of CYP3A, P-gp and UGT1A1 together (i.e., all 3 pathways), such as atazanavir/cobicistat, may significantly increase plasma concentrations of lenacapavir, therefore co-administration is not recommended (see section 4.4).
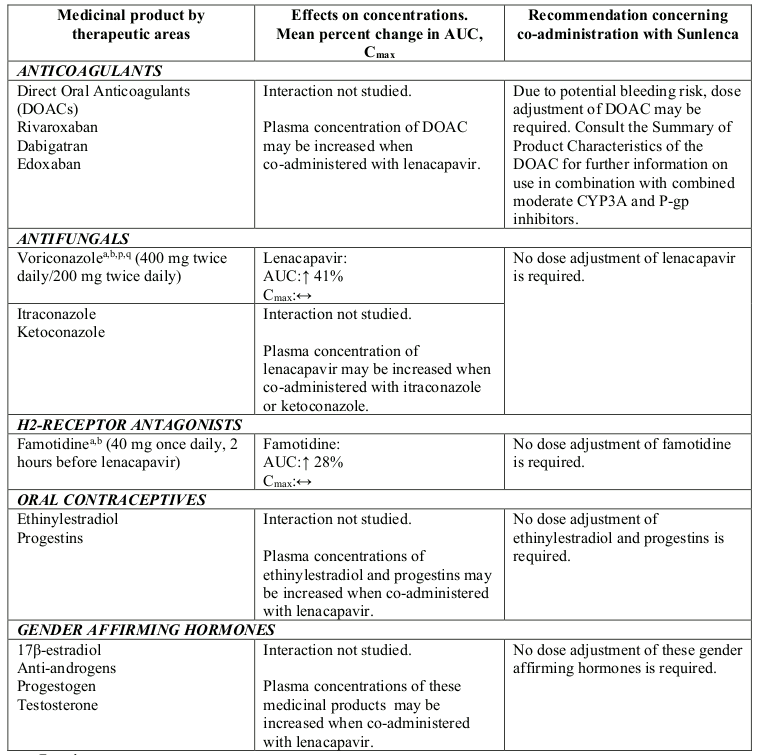
Strong CYP3A4 inhibitors alone (e.g. voriconazole) or strong inhibitors of CYP3A4 and P-gp together (e.g. cobicistat) do not result in a clinically meaningful increase in lenacapavir exposures.
Effect of lenacapavir on the pharmacokinetics of other medicinal products
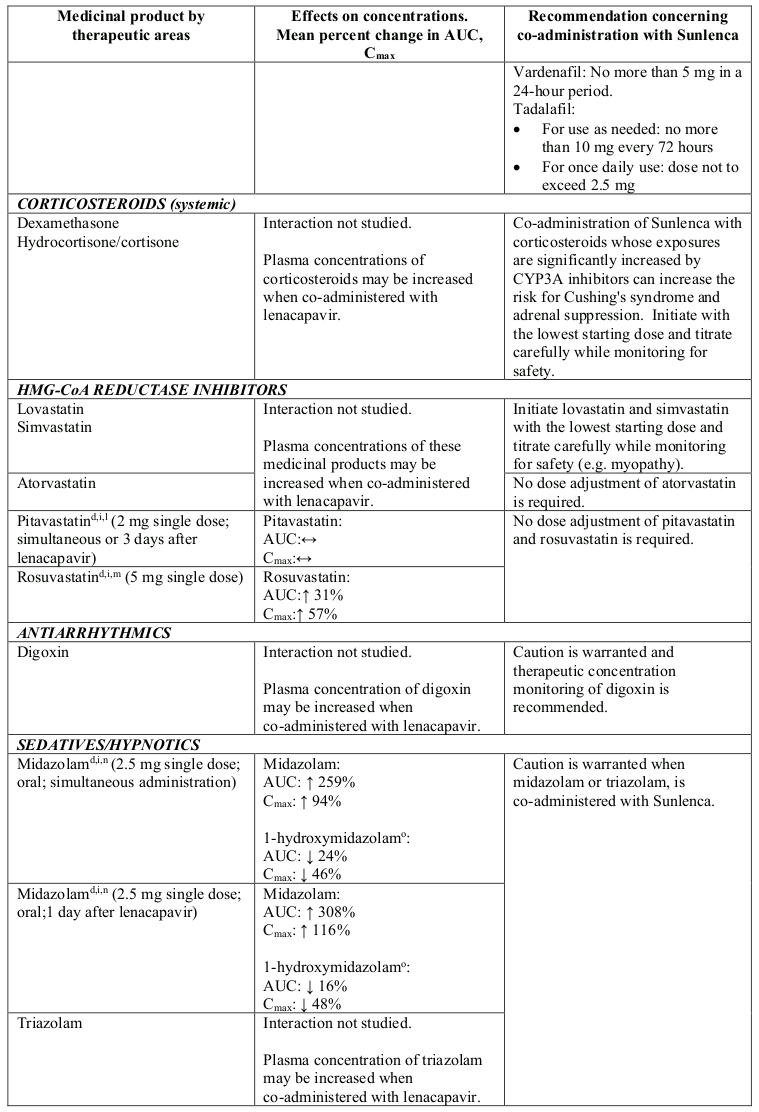
Lenacapavir is a moderate inhibitor of CYP3A. Caution is advised if Sunlenca is co-administered with a sensitive CYP3A substrate with a narrow therapeutic index. Lenacapavir is not a clinically meaningful inhibitor of P-gp and BCRP and does not inhibit OATP.
Table 2. Interactions between Sunlenca and other medicinal products:
a Fasted.
b This study was conducted using lenacapavir 300 mg single dose administered orally.
c Evaluated as a strong inducer of CYP3A, and an inducer of P-gp and UGT.
d Fed.
e Evaluated as a strong inhibitor of CYP3A, and an inhibitor UGT1A1 and P-gp.
f Evaluated as a moderate inducer of CYP3A and an inducer of P-gp.
g Evaluated as a strong inhibitor of CYP3A and an inhibitor of P-gp.
h Evaluated as a strong inhibitor of CYP3A, and an inhibitor and inducer of P-gp.
i This study was conducted using lenacapavir 600 mg single dose following a loading regimen of 600 mg twice daily for 2 days, single 600 mg doses of lenacapavir were administered with each co-administered medicinal product.
j Evaluated as a P-gp substrate.
k Tenofovir alafenamide is converted to tenofovir in vivo.
l Evaluated as an OATP substrate.
m Evaluated as an BCRP substrate.
n Evaluated as a CYP3A substrate.
° Major active metabolite of midazolam.
p Evaluated as a strong inhibitor of CYP3A.
q This study was conducted using voriconazole 400 mg loading dose twice daily for a day, followed by 200 mg maintenance dose twice daily.
4.6. Fertility, pregnancy and lactation
Pregnancy
There are no or limited amount of data from the use of lenacapavir in pregnant women.
Animal studies do not indicate direct or indirect harmful effects with respect to pregnancy, foetal development, parturition or postnatal development (see section 5.3).
As a precautionary measure, it is preferable to avoid the use of Sunlenca during pregnancy unless the clinical condition of the women requires treatment with Sunlenca.
Breast-feeding
In order to avoid transmission of HIV to the infant it is recommended that HIV-infected women do not breast-feed their infants.
It is unknown whether lenacapavir is excreted in human milk. After administration to rats during pregnancy and lactation, lenacapavir was detected at low levels in the plasma of nursing rat pups, without effects on these nursing pups.
Fertility
There are no data on the effects of lenacapavir on human male or female fertility. Animal studies indicate no effects on lenacapavir on male or female fertility (see section 5.3).
4.7. Effects on ability to drive and use machines
Sunlenca is expected to have no or negligible influence on the ability to drive and use machines.
4.8. Undesirable effects
Summary of the safety profile
The most common adverse reactions in heavily treatment experienced adult patients with HIV were injection site reactions (ISRs) (63%) and nausea (4%).
Tabulated list of adverse reactions
A tabulated list of adverse reactions is presented in Table 3. Frequencies are defined as very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000), and not known (cannot be estimated from the available data).
Table 3. Tabulated list of adverse reactions:
| Frequencya | Adverse reaction |
|---|---|
| Immune system disorders | |
| Not known | immune reconstitution inflammatory syndrome |
| Gastrointestinal disorders | |
| Common | nausea |
| General disorders and administration site conditions | |
| Very common | injection site reactionsb |
a Frequency based on all patients (Cohorts 1 and 2) in CAPELLA (see section 5.1).
b Includes injection site swelling, pain, nodule, erythema, induration, pruritus, extravasation, discomfort, mass, haematoma, oedema, and ulcer.
Description of selected adverse reactions
Immune Reconstitution Inflammatory Syndrome
In HIV infected patients with severe immune deficiency at the time of initiation of CART, an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
Local injection site reactions
Most patients had ISRs that were mild (Grade 1, 42%) or moderate (Grade 2, 18%). Three percent of patients experienced a severe (Grade 3) ISR that resolved within 1 to 8 days. No patients experienced a Grade 4 ISR. The median duration of all ISRs excluding nodules and indurations was 6 days. The median duration of nodules and indurations was 180 and 118 days, respectively.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
6.2. Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.