TALTZ Solution for injection Ref.[8982] Active ingredients: Ixekizumab
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Eli Lilly and Company (Ireland) Limited, Dunderrow, Kinsale, Co. Cork, Ireland
Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, interleukin inhibitors
ATC code: L04AC13
Mechanism of action
Ixekizumab is an IgG4 monoclonal antibody that binds with high affinity (<3 pM) and specificity to interleukin 17A (both IL-17A and IL-17A/F). Elevated concentrations of IL-17A have been implicated in the pathogenesis of psoriasis by promoting keratinocyte proliferation and activation, as well as in the pathogenesis of psoriatic arthritis and axial spondyloarthritis by driving inflammation leading to erosive bone damage and pathological new bone formation. Neutralisation of IL-17A by ixekizumab inhibits these actions. Ixekizumab does not bind to ligands IL-17B, IL-17C, IL-17D, IL-17E or IL-17F.
In vitro binding assays confirmed that ixekizumab does not bind to human Fcγ receptors I, IIa, and IIIa or to complement component C1q.
Pharmacodynamic effects
Ixekizumab modulates biological responses that are induced or regulated by IL-17A. Based on psoriatic skin biopsy data from a phase I study, there was a dose-related trend towards decreased epidermal thickness, number of proliferating keratinocytes, T cells, and dendritic cells, as well as reductions in local inflammatory markers from baseline to day 43. As a direct consequence treatment with ixekizumab reduces erythema, induration and desquamation present in plaque psoriasis lesions.
Ixekizumab has been shown to lower (within 1 week of treatment) levels of C-reactive protein, which is a marker of inflammation.
Clinical efficacy and safety
Adult plaque psoriasis
The efficacy and safety of ixekizumab were assessed in three randomised, double-blind, placebo-controlled phase III studies in adult patients (N=3 866) with moderate to severe plaque psoriasis who were candidates for phototherapy or systemic therapy (UNCOVER-1, UNCOVER-2, and UNCOVER-3). The efficacy and safety of ixekizumab were also evaluated versus etanercept (UNCOVER-2 and UNCOVER-3). Patients randomised to ixekizumab who were sPGA (0,1) responders (static Physicians Global Assessment) at week 12 were re-randomised to receive placebo or ixekizumab for an additional 48 weeks (UNCOVER-1 and UNCOVER-2); patients randomised to placebo, etanercept or ixekizumab who were sPGA (0,1) non-responders received ixekizumab for up to 48 weeks. In addition, long-term efficacy and safety were evaluated in all three studies for up to a total of 5 years in patients who participated through the entire study.
64% of patients had received prior systemic therapy (biologic, conventional systemic or psoralen and ultraviolet A (PUVA)), 43.5% prior phototherapy, 49.3% prior conventional systemic therapy, and 26.4% prior biologic therapy. 14.9% had received at least one anti-TNF alpha agent, and 8.7% an anti-IL-12/IL-23. 23.4% of patients had a history of psoriatic arthritis at baseline.
In all three studies, the co-primary endpoints were the proportion of patients who achieved a PASI 75 response (Psoriasis Area and Severity Index) and an sPGA of “0” (“clear”) or 1 (“minimal”) response at week 12 versus placebo. The median baseline PASI score ranged from 17.4 to 18.3; 48.3% to 51.2% of patients had a baseline sPGA score of severe or very severe, and mean baseline itch
Numeric Rating Scale (itch NRS) ranged from 6.3 to 7.1.
Clinical response at 12 weeks
UNCOVER-1 randomised 1 296 patients (1:1:1) to receive either placebo or ixekizumab (80 mg every two or four weeks [Q2W or Q4W] following a 160 mg starting dose) for 12 weeks.
Table 2. Efficacy results at week 12 in UNCOVER-1:
| Endpoints | Number of patients (%) | Difference from placebo in response rate (95% CI) | |||
|---|---|---|---|---|---|
| Placebo (N=431) | Ixekizumab 80 mg Q4W (N=432) | Ixekizumab 80 mg Q2W (N=433) | Ixekizumab 80 mg Q4W | Ixekizumab 80 mg Q2W | |
| sPGA of “0” (clear) or “1” (minimal) | 14 (3.2) | 330 (76.4)a | 354 (81.8)a | 73.1 (68.8, 77.5) | 78.5 (74.5, 82.5) |
| sPGA of “0” (clear) | 0 | 149 (34,5)a | 160 (37.0)a | 34.5 (30.0, 39.0) | 37.0 (32.4, 41.5) |
| PASI 75 | 17 (3,9) | 357 (82,6)a | 386 (89.1)a | 78.7 (74.7, 82.7) | 85.2 (81.7, 88.7) |
| PASI 90 | 2 (0.5) | 279 (64.6)a | 307 (70.9)a | 64.1 (59.6, 68.7) | 70.4 (66.1, 74.8) |
| PASI 100 | 0 | 145 (33,6)a | 153 (35.3)a | 33.6 (29.1, 38.0) | 35.3 (30.8, 39.8) |
| Itch NRS reduction ≥4b | 58 (15.5) | 305 (80.5)a | 336 (85.9)a | 65.0 (59.5, 70.4) | 70.4 (65.4, 75.5) |
Abbreviations: N = number of patients in the intent-to-treat population
Note: patients with missing data were counted as non-responders
a p<0.001 compared with placebo
b Patients with Itch NRS ≥4 at baseline: placebo N=374, ixekizumab 80 mg Q4W N=379, ixekizumab 80 mg Q2W N=391
UNCOVER-2 enrolled 1,224 patients. Patients were randomised (1:2:2:2) to receive either placebo, or ixekizumab (80 mg every two or four weeks [Q2W or Q4W] following a 160 mg starting dose) or etanercept 50 mg twice weekly for 12 weeks.
Table 3. Efficacy results at week 12 in UNCOVER-2:
| Endpoints | Number of patients (%) | Difference from placebo in response rate (95% CI) | ||||
|---|---|---|---|---|---|---|
| Placebo (N=168) | Ixekizumab 80 mg Q4W (N=347) | Ixekizumab 80 mg Q2W (N=351) | Etanercept 50 mg twice weekly (N=358) | Ixekizumab 80 mg Q4W | Ixekizumab 80 mg Q2W | |
| sPGA of “0” (clear) or "1" (minimal) | 4 (2.4) | 253 (72.9)a | 292 (83.2)a | 129 (36.0) | 70.5 (65.3, 75.7) | 80.8 (76.3, 85.4) |
| sPGA of “0” (clear) | 1 (0.6) | 112 (32.3)a,b | 147 (41,9)a,b | 21 (5.9)c | 31.7 (26.6, 36.7) | 41.3 (36.0, 46.6) |
| PASI 75 | 4 (2.4) | 269 (77.5)a,b | 315 (89.7)a,b | 149 (41.6)a | 75.1 (70.2, 80.1) | 87.4 (83.4, 91.3) |
| PASI 90 | 1 (0.6) | 207 (59,7)a,b | 248 (70.7)a,b | 67 (18.7)a | 59.1 (53.8, 64.4) | 70.1 (65.2, 75.0) |
| PASI 100 | 1 (0.6) | 107 (30.8)a,b | 142 (40.5)a,b | 19 (5.3)c | 30.2 (25.2, 35.2) | 39.9 (34.6, 45.1) |
| Itch NRS reduction ≥4d | 19 (14.1) | 225 (76.8)a,b | 258 (85.1)a,b | 177 (57.8)a | 62.7 (55.1, 70.3) | 71.1 (64.0, 78.2) |
Abbreviations: N = number of patients in the intent-to-treat population
Note: patients with missing data were counted as non-responders.
a p<0.001 compared with placebo
b p<0.001 compared with etanercept
c p<0.01 compared with placebo
d Patients with Itch NRS ≥4 at baseline: placebo N=135, ixekizumab 80 mg Q4W N=293, ixekizumab 80 mg Q2W N=303, Etanercept N=306
UNCOVER-3 randomised 1 346 patients (1:2:2:2) to receive either placebo, or ixekizumab (80 mg every two or four weeks [Q2W or Q4W] following a 160 mg starting dose) or etanercept 50 mg twice weekly for 12 weeks.
Table 4. Efficacy results at week 12 in UNCOVER-3:
| Endpoints | Number of patients (%) | Difference from placebo in response rate (95% CI) | ||||
|---|---|---|---|---|---|---|
| Placebo (N=193) | Ixekizumab 80 mg Q4W (N=386) | Ixekizumab 80 mg Q2W (N=385) | Etanercept 50 mg twice weekly (N=382) | Ixekizumab 80 mg Q4W | Ixekizumab 80 mg Q2W | |
| sPGA of “0” (clear) or "1" (minimal) | 13 (6.7) | 291 (75.4)a,b | 310 (80.5)a,b | 159 (41.6)a | 68.7 (63.1, 74.2) | 73.8 (68.5, 79.1) |
| sPGA of “0” (clear) | 0 | 139 (36.0)a,b | 155 (40.3)a,b | 33 (8.6)a | 36.0 (31.2, 40.8) | 40.3 (35.4, 45.2) |
| PASI 75 | 14 (7.3) | 325 (84.2)a,b | 336 (87.3)a,b | 204 (53.4)a | 76.9 (71.8, 82.1) | 80.0 (75.1, 85.0) |
| PASI 90 | 6 (3.1) | 252 (65.3)a,b | 262 (68.1)a,b | 98 (25,7)a | 62.2 (56.8, 67.5) | 64.9 (59.7, 70.2) |
| PASI 100 | 0 | 135 (35.0)a,b | 145 (37.7)a,b | 28 (7.3)a | 35 (30.2, 39.7) | 37.7 (32.8, 42.5) |
| Itch NRS reduction ≥4c | 33 (20.9) | 250 (79.9)a,b | 264 (82.5)a,b | 200 (64.1)a | 59.0 (51.2, 66.7) | 61.6 (54.0, 69.2) |
Abbreviations: N = number of patients in the intent-to-treat population
Note: patients with missing data were counted as non-responders
a p<0.001 compared with placebo
b p<0.001 compared with etanercept
c Patients with Itch NRS ≥4 at baseline: placebo N=158, ixekizumab 80 mg Q4W N=313, ixekizumab 80 mg Q2W N=320, etanercept N=312
Ixekizumab was associated with a fast onset of efficacy with >50% reduction in mean PASI by Week 2 (Figure 1). The percentage of patients achieving PASI 75 was significantly greater for ixekizumab compared with placebo and etanercept as early as Week 1. Approximately 25% of patients treated with ixekizumab achieved a PASI score <5 by Week 2, more than 55% achieved the PASI score <5 by Week 4, and increased to 85% by Week 12 (compared to 3%, 14% and 50% for etanercept). Significant improvements in itch severity were seen at Week 1 in patients treated with ixekizumab.
Figure 1. PASI score, percent improvement at each post baseline visit (mBOCF)) in the intent-to-treat population during the induction dosing period – UNCOVER-2 and UNCOVER-3:

The efficacy and safety of ixekizumab was demonstrated regardless of age, gender, race, body weight, PASI baseline severity, plaques location, concurrent psoriatic arthritis, and previous treatment with a biologic. Ixekizumab was efficacious in systemic treatment-naive, biologic-naive, biologic/anti-TNF-exposed and biologic/anti-TNF-failure patients.
For patients identified as an sPGA (0,1) non-responder to etanercept at week 12 in UNCOVER-2 (N=200) and who were switched to ixekizumab 80 mg Q4W after a 4 week washout period, 73% and 83.5% of patients achieved sPGA (0,1) and PASI 75, respectively, after 12 weeks of treatment with ixekizumab.
In the 2 clinical studies that included an active comparator (UNCOVER-2 and UNCOVER-3), the rate of serious adverse events was 1.9% for both etanercept and for ixekizumab, and the rate of discontinuation due to adverse events was 1.2% for etanercept and 2.0% for ixekizumab. The rate of infections was 21.5% for etanercept and 26.0% for ixekizumab, with 0.4% being serious for etanercept and 0.5% for ixekizumab.
Maintenance of response at week 60 and up to 5 years
Patients originally randomised to ixekizumab and who were responders at week 12 (i.e., sPGA score of 0,1) in UNCOVER-1 and UNCOVER-2 were re-randomised to an additional 48 weeks of treatment with placebo or ixekizumab (80 mg every four or twelve weeks [Q4W or Q12W]).
For sPGA (0,1) responders at week 12 re-randomised to treatment withdrawal (i.e., placebo), the median time to relapse (sPGA ≥3) was 164 days in integrated UNCOVER-1 and UNCOVER-2 studies. Among these patients, 71.5% regained at least an sPGA (0,1) response within 12 weeks of restarting treatment with ixekizumab 80 mg Q4W.
Table 5. Maintenance of response and efficacy at week 60 (Studies UNCOVER-1 and UNCOVER-2):
| Endpoints | Number of patients (%) | Difference from placebo in response rate (95% CI) | ||||
|---|---|---|---|---|---|---|
| 80 mg Q4W (induction) / Placebo (maintenance) (N=191) | 80 mg Q2W (induction) / Placebo (maintenance) (N=211) | 80 mg Q4W (induction) / 80 mg Q4W (maintenance) (N=195) | 80 mg Q2W (induction) / 80 mg Q4W (maintenance) (N=221) | 80 mg Q4W (induction) / 80 mg Q4W (maintenance) | 80 mg Q2W (induction) / 80 mg Q4W (maintenance) | |
| Maintained sPGA of “0” (clear) or “1” (minimal) | 12 (6.3) | 16 (7.6) | 134 (68.7)a | 173 (78.3)a | 62.4 (55.1, 69.8) | 70.7 (64.2, 77.2) |
| Maintained or achieved sPGA “0” (clear) | 3 (1.6) | 6 (2.8) | 96 (49.2)a | 130 (58.8)a | 47.7 (40.4, 54.9) | 56.0 (49.1, 62.8) |
| Maintained or achieved PASI 75 | 15 (7.9) | 19 (9.0) | 145 (74.4)a | 184 (83.3)a | 66.5 (59.3, 73.7) | 74.3 (68.0, 80.5) |
| Maintained or achieved PASI 90 | 9 (4.7) | 10 (4.7) | 130 (66.7)a | 169 (76.5)a | 62.0 (54.7, 69.2) | 71.7 (65.4, 78.0) |
| Maintained or achieved PASI 100 | 3 (1.6) | 6 (2.8) | 97 (49.7)a | 127 (57.5)α | 48.2 (40.9, 55.4) | 54.6 (47.7, 61.5) |
Abbreviations: N = number of patients in the analysis population
Note: patients with missing data were counted as non-responders
a p<0.001 compared with placebo
Ixekizumab was efficacious in the maintenance of response in systemic treatment-naive, biologic-naive, biologic/anti-TNF-exposed and biologic/anti-TNF-failure patients.
Significantly greater improvements at week 12 from baseline compared to placebo and etanercept were demonstrated in nail psoriasis (as measured by the Nail Psoriasis Severity Index [NAPSI]), in scalp psoriasis (as measured by Psoriasis Scalp Severity Index [PSSI]) and in palmoplantar psoriasis (as measured by Psoriasis Palmoplantar Severity Index [PPASI]) and were maintained at week 60 in patients treated with ixekizumab who were sPGA (0,1) responders at week 12.
Of 591 subjects who received ixekizumab Q2W during the Induction Period then Q4W afterward in study UNCOVER-1, UNCOVER-2, and UNCOVER-3, 427 subjects completed 5 years of ixekizumab treatment, among those 101 patients required a dose escalation. Among the patients who completed the week 264 assessment (N=427), 295 patients (69%), 289 patients (68%) and 205 patients (48%) were observed to have sPGA (0,1), PASI 90 and PASI 100 response, respectively, at week 264. DLQI were collected after Induction Period in UNCOVER-1 and UNCOVER-2, 113 patients (66%) were observed to have DLQI (0,1) response.
Quality of life/patient-reported outcomes
At week 12 and across studies, ixekizumab was associated with statistically significant improvement in Health-related Quality of Life as assessed by mean decrease ranges from baseline in the Dermatology Life Quality Index (DLQI) (ixekizumab 80 mg Q2W from -10.2 to -11.1, ixekizumab 80 mg Q4W from -9.4 to -10.7, etanercept from -7.7 to -8.0 and placebo -1.0 to -2.0). A significantly greater proportion of patients treated with ixekizumab achieved a DLQI 0 or 1. Across studies a significantly greater proportion of patients treated with ixekizumab achieved a reduction of Itch NRS ≥4 points at week 12 (84.6% for ixekizumab Q2W, 79.2% for ixekizumab Q4W and 16.5% for placebo) and the benefit was sustained over time up to week 60 in patients treated with ixekizumab who were sPGA (0 or 1) responders at week 12. There was not any evidence of worsening of depression up to 60 weeks treatment with ixekizumab as assessed by the Quick Inventory of Depressive Symptomatology Self Report.
Post-marketing direct comparative studies
IXORA-S: In a double-blind study ixekizumabwas superior against ustekinumab on the primary study objective PASI 90 response at week 12 (Table 6). Onset of response was superior on PASI 75 as early as week 2 (p<0.001) and on PASI 90 and PASI 100 by week 4 (p<0.001). Superiority of ixekizumab versus ustekinumab was also demonstrated in the subgroups stratified by weight.
Table 6. PASI-response rates from comparative study ixekizumab versus ustekinumab:
| Week 12 | Week 24 | Week 52 | ||||
|---|---|---|---|---|---|---|
| Ixekizumab* | Ustekinumab** | Ixekizumab* | Ustekinumab** | Ixekizumab* | Ustekinumab** | |
| Patients (n) | 136 | 166 | 136 | 166 | 136 | 166 |
| PASI 75, n (%) | 120 (88.2%) | 114 (68.7%) | 124 (91.2%) | 136 (81.9%) | 120 (88.2%) | 126 (75.9%) |
| PASI 90, n (%) | 99 (72.8%)§ | 70 (42.2%) | 113 (83.1%) | 98 (59.0%) | 104 (76.5%) | 98 (59.0%) |
| PASI 100, n (%) | 49 (36.0%) | 24 (14.5%) | 67 (49.3%) | 39 (23.5%) | 71 (52.2%) | 59 (35.5%) |
* Ixekizumab 160 mg was given as a loading dose followed by 80 mg at week 2,4,6,8,10 and 12, and 80 mg Q4W thereafter
** Weight based dosing: Patients treated with ustekinumab received 45 mg or 90 mg at Weeks 0 and 4, then every 12 weeks until Week 52 (dosed by weight as per approved posology)
§ p<0.001 versus ustekinumab (p value only provided for primary endpoint)
IXORA-R: Efficacy and safety of ixekizumab was also investigated in a 24-week randomised, double-blind, parallel-group study comparing ixekizumab to guselkumab, with ixekizumab being superior as early as week 4 in achieving complete skin clearance and on the primary study objective (PASI 100 at week 12) and non-inferior on PASI 100 at week 24 (Table 7).
Table 7. Efficacy Responses from comparative study ixekizumab versus guselkumab, Intent-to-Treat Populationa:
| Endpoint | Time point | Guselkulmab (N=507) response, n (%) | Ixekizumab (N=520) response, n (%) | Difference (IXE – GUS), % (CI) | p-value |
|---|---|---|---|---|---|
| Primary Objective | |||||
| PASI 100 | Week 12 | 126 (24.9) | 215 (41.3) | 16.5 (10.8, 22.2) | <0.001 |
| Major Secondary Objectives | |||||
| PASI 75 | Week 2 | 26 (5.1) | 119 (22.9) | 17.8 (13.7, 21.8) | <0.001 |
| PASI 90 | Week 4 | 40 (7.9) | 109 (21.0) | 13.1 (8.9, 17.3) | <0.001 |
| PASI 100 | Week 4 | 7 (1.4) | 35 (6.7) | 5.4 (3.0, 7.7) | <0.001 |
| PASI 90 | Week 8 | 182 (35.9) | 304 (58.5) | 22.6 (16.6, 28.5) | <0.001 |
| sPGA (0) | Week 12 | 128 (25.2) | 218 (41.9) | 16.7 (11.0, 22.4) | <0.001 |
| PASI 50 | Week 1 | 47 (9.3) | 143 (27.5) | 18.2 (13.6, 22.8) | <0.001 |
| PASI 100 | Week 8 | 69 (13.6) | 154 (29.6) | 16.0 (11.1, 20.9) | <0.001 |
| PASI 100 | Week 24 | 265 (52.3) | 260 (50.0) | -2.3 (-8.4, 3.8) | 0.414 |
Abbreviations: CI = confidence interval; GUS = guselkumab; IXE = ixekizumab; N = number of patients in the analysis population; n = number of patients in the specified category; PASI = psoriasis area and severity index; sPGA = static physician global assessment.
a Endpoints were gated in this order
Figure 2. PASI 100 at weeks 4, 8, 12 and 24, NRI:

* p<0.001 vs guselkumab at weeks 4, 8, and 12
NRI = Non-Responder Imputation
Efficacy in genital psoriasis
A randomised, double-blind, placebo-controlled study (IXORA-Q) was conducted in 149 adult subjects (24% females) with moderate to severe genital psoriasis (sPGA of Genitalia score of ≥3), a minimum body surface area (BSA) involvement of 1% (60.4% had a BSA ≥10%) and previous failure of or intolerance to at least one topical therapy for genital psoriasis. Patients had at least moderate plaque psoriasis (defined as sPGA score of ≥3 and being candidates for phototherapy and/or systemic therapy) for at least 6 months.
Subjects randomised to ixekizumab received an initial dose of 160 mg followed by 80 mg every 2 weeks for 12 weeks. The primary endpoint was the proportion of patients who achieved at least a “0” (clear) or “1” (minimal) response on the sPGA of Genitalia (sPGA of Genitalia 0/1). At week 12, significantly more subjects in the ixekizumab group than placebo group achieved a sPGA of Genitalia 0/1 and a sPGA 0/1 independent of baseline BSA (baseline BSA 1% - <10% resp. ≥10%: sPGA of Genitalia “0” or “1”: ixekizumab 71%, resp. 75%; placebo: 0%, resp. 13%). A significantly greater proportion of patients treated with ixekizumab achieved a reduction in the PROs of severity of genital pain, genital itch, impact of genital psoriasis on sexual activity, and Dermatology Quality of Life Index (DLQI).
Table 8. Efficacy results at week 12 in Adults with genital psoriasis in trial IXORA-Q; NRIa:
| Endpoints | Ixekizumab | Placebo | Difference from placebo (95% CI) |
|---|---|---|---|
| Number of patients (N) randomized | N=75 | N=74 | |
| sPGA of Genitalia “0” or “1” | 73% | 8% | 65% (53%, 77%) |
| sPGA “0” ή “1” | 73% | 3% | 71% (60%, 81%) |
| DLQI 0,1b | 45% | 3% | 43% (31%, 55%) |
| N with baseline GPSS Itch NRS Score ≥3 | N=62 | N=60 | |
| GPSS Genital Itch (≥3 point improvement) | 60% | 8% | 51% (37%, 65%) |
| N with baseline SFQ Item 2 Score ≥2 | N=37 | N=42 | |
| SFQ-item 2 score, “0” (never limited) or “1” (rarely limited) | 78% | 21% | 57% (39%, 75%) |
a Abbreviations: NRI = Non-Responder Imputation; sPGA = static Physician Global Assessment; GPSS = Genital Psoriasis Symptom Scale; SFQ = Sexual Frequency Questionnaire; DLQI = Dermatology Quality of Life Index; bTotal DLQI score of 0,1 indicates skin condition has no effect at all on patient’s life. sPGA of “0” or “1” is equivalent to “clear” or “minimal”; NRS = Numeric Rating Scale
Paediatric plaque psoriasis
A randomised, double-blind, multicenter, placebo-controlled trial (IXORA-Peds) enrolled 201 children 6 to less than 18 years of age, with moderate to severe plaque psoriasis (as defined by a sPGA score ≥3, involving ≥10% of the body surface area, and a PASI score ≥12) who were candidates for phototherapy or systemic therapy, or were inadequately controlled on topical therapy. Patients were randomised to placebo (n=56), etanercept (n=30) or ixekizumab (n=115) with dosing stratified by weight:
- <25 kg: 40 mg at week 0 followed by 20 mg Q4W (n=4)
- 25 kg to 50 kg: 80 mg at week 0 followed by 40 mg Q4W (n=50)
- >50 kg: 160 mg at week 0 followed by 80 mg Q4W (n=147)
Patients randomised to etanercept (patients with severe psoriasis) received 0.8 mg/kg, not exceeding 50 mg per dose, every week from week 0 through week 11.
Response to treatment was assessed after 12 weeks and defined by the proportion of patients who achieved the co-primary endpoint of an sPGA score of “0” (clear) or “1” (almost clear) with at least a 2 point improvement from baseline and the proportion of patients that achieved a reduction in PASI score of at least 75% (PASI 75) from baseline.
Other evaluated outcomes at week 12 included the proportion of patients who achieved PASI 90, PASI 100, sPGA of “0” and an improvement of itch severity as measured by a reduction of at least 4 points on an 11-point itch Numeric Rating Scale.
Patients had a median baseline PASI of 17 score ranging from 12-49. Baseline sPGA score was severe or very severe in 49%. Of all patients, 22% had received prior phototherapy and 32% had received prior conventional systemic therapy for the treatment of psoriasis. 25% of patients (n=43) were below 12 years (14% of patients [n=24] were 6-9 years and 11% of patients [n=19] were 10-11 years); 75% (n=128) were 12 years or above.
The clinical response data are presented in Table 9.
Table 9. Efficacy results in pediatric patients with plaque psoriasis, NRI:
| Endpoints | Ixekizumaba (N=115) n (%) | Placebo (N=56) n (%) | Difference vs placebo (95% CI) | Etanerceptb (N=30) n (%) | Difference vs etanercept (95% CI)b |
|---|---|---|---|---|---|
| sPGA “0” (clear) or “1” (almost clear)c | |||||
| week 4 | 55 (48) | 4 (7) | 40.7 (29.3, 52.0)f | 0(0) | 36.8 (21.5, 52.2) |
| week 12c | 93 (81) | 6 (11) | 70.2 (59.3, 81.0)f | 16 (53) | 23.0 (0.6, 45.4) |
| sPGA “0” (clear)d | 60 (52) | 1 (2) | 50.4 (40.6, 60.2)f | 5 (17) | 46.5 (26.2, 66.8) |
| PASI 75 | |||||
| week 4 | 62 (54) | 5 (9) | 45.0 (33.2, 56.8)f | 3 (10) | 34.7 (15.6, 53.8) |
| week 12c | 102 (89) | 14 (25) | 63.7 (51.0, 76.4)f | 19 (63) | 20.9 (0.1, 41.7) |
| PASI 90d | 90 (78) | 3 (5) | 72.9 (63.3, 82.5)f | 12 (40) | 36.3 (14.2, 58.5) |
| PASI 100d | 57 (50) | 1 (2) | 47.8 (38.0, 57.6)f | 5 (17) | 43.9 (23.4, 64.3) |
| Itch NRS (≥4 point improvement)d,e | 59 (71) | 8 (20) | 51.1 (35.3, 66.9)f | Not evaluated | --- |
Abbreviations: N = Number of patients in the intent-to-treat population; NRI = Non-Responder Imputation.
a At week 0, subjects received 160 mg, 80 mg, or 40 mg of ixekizumab, followed by 80 mg, 40 mg, or 20 mg every 4 weeks, depending on weight category, for 12 weeks.
b Comparisons to etanercept were performed within the sub-population of patients outside of US and Canada with severe Ps (N for ixekizumab = 38).
c Co-primary endpoints.
d Results at week 12.
^^e Itch NRS (≥4 improvement) in patients with baseline Itch NRS ≥4. The number of ITT patients with baseline Itch NRS Score ≥4 are as follows: ixekizumab, n=83; PBO, n=40.
f p<0.001
Figure 3. Percent of patients achieving PASI 75 in pediatric psoriasis through week 12:

Patients in the ixekizumab treatment group had clinically meaningful higher CDLQI/DLQI (0,1) responses at week 12 (NRI) compared with placebo. The difference between treatment groups was apparent from as early as week 4.
There were greater improvements at week 12 from baseline compared to placebo in nail psoriasis (as measured by the Nail Psoriasis Severity Index [NAPSI=0: ixekizumab 18% (6/34), placebo 0% (0/12)]), in scalp psoriasis (as measured by Psoriasis Scalp Severity Index [PSSI=0: ixekizumab 69% (70/102), placebo 16% (8/50)]) and in palmoplantar psoriasis (as measured by Psoriasis Palmoplantar Severity Index [PPASI 75: ixekizumab 53% (9/17), placebo 11% (1/9)]).
Psoriatic arthritis
Ixekizumab was assessed in two randomised, double-blind, placebo-controlled phase III studies in 780 patients with active psoriatic arthritis (≥3 swollen and ≥3 tender joints). Patients had a diagnosis of psoriatic arthritis (Classification Criteria for Psoriatic Arthritis [CASPAR] criteria) for a median of 5.33 years and had current plaque psoriasis skin lesions (94.0%) or a documented history of plaque psoriasis, with 12.1% of patients with moderate to severe plaque psoriasis at baseline. Over 58.9% and 22.3% of the psoriatic arthritis patients had enthesitis and dactylitis at baseline, respectively. Primary endpoint of both studies was American College of Rheumatology (ACR) 20 response at week 24, followed by a long-term extension period from week 24 to week 156 (3 years).
In Psoriatic Arthritis Study 1 (SPIRIT-P1), patients naive to biologic therapy with active psoriatic arthritis were randomised to placebo, adalimumab 40 mg once every 2 weeks (active control reference arm), ixekizumab 80 mg once every 2 weeks (Q2W), or 80 mg once every 4 weeks (Q4W). Both ixekizumab regimens included a 160 mg starting dose. 85.3% of patients in this study had received prior treatment with ≥1 cDMARD. 53% of patients had concomitant use of MTX at a mean weekly dose of 15.8 mg. 67% of patients who had concomitant use of MTX had a dose of 15 mg or greater. Patients with an inadequate response at week 16 received rescue therapy (modification to background therapy). Patients on ixekizumab Q2W or Q4W remained on their originally assigned dose of ixekizumab. Patients receiving adalimumab or placebo were re-randomised 1:1 to ixekizumab Q2W or Q4W at week 16 or 24 based on responder status. 243 patients completed the extension period of 3 years on ixekizumab.
Psoriatic Arthritis Study 2 (SPIRIT-P2) enrolled patients who were previously treated with an anti-TNF agent and discontinued the anti-TNF agent for either lack of efficacy or intolerance (anti-TNF-IR patients). Patients were randomised to placebo, ixekizumab 80 mg once every 2 weeks (Q2W), or 80 mg once every 4 weeks (Q4W). Both ixekizumab regimens included a 160 mg starting dose. 56% and 35% of patients were inadequate responders to 1 anti-TNF or 2 anti-TNF, respectively. SPIRIT-P2 evaluated 363 patients, of whom 41% had concomitant use of MTX at a mean weekly dose of 16.1 mg. 73.2% of patients who had concomitant use of MTX had a dose of 15 mg or greater. Patients with an inadequate response at week 16 received rescue therapy (modification to background therapy). Patients in ixekizumab Q2W or Q4W remained on their originally assigned dose of ixekizumab. Patients receiving placebo were re-randomised 1:1 to ixekizumab Q2W or Q4W at week 16 or 24 based on responder status. 168 patients completed the extension period of 3 years on ixekizumab.
Signs and symptoms
Treatment with ixekizumab resulted in significant improvement in measures of disease activity compared to placebo at week 24 (see Table 10)
Table 10. Efficacy results in SPIRIT-P1 and SPIRIT-P2 at week 24:
| SPIRIT-P1 | SPIRIT-P2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Endpoints | Difference from placebo in response rate (95% CI) | Difference from placebo in response rate (95% CI) | |||||||||
| PBO (N=106) | Ixekizumab Q4W (N=107) | Ixekizumab Q2W (N=103) | ADA (N=101) | Ixekizumab Q4W | Ixekizumab Q2W | PBO (N=118) | Ixekizumab Q4W (N=122) | Ixekizumab Q2W (N=123) | Ixekizumab Q4W | Ixekizumab Q2W | |
| ACR 20 response, n (%) | |||||||||||
| Week 24 | 32 (30.2) | 62 (57.9) | 64 (62.1) | 58 (57.4) | 27.8 (15.0, 40.6)c | 31.9 (19.1, 44.8)c | 23 (19.5) | 65 (53.3) | 59 (48.0) | 33.8 (22.4, 45.2)c | 28.5 (17.1, 39.8)c |
| ACR 50 response, n (%) | |||||||||||
| Week 24 | 16 (15.1) | 43 (40.2) | 48 (46.6) | 39 (38.6) | 25.1 (13.6, 36.6)c | 31.5 (19.7, 43.3)c | 6 (5.1) | 43 (35.2) | 41 (33.3) | 30.2 (20.8, 39.5)c | 28.3 (19.0, 37.5)c |
| ACR 70 response, n (%) | |||||||||||
| Week 24 | 6 (5.7) | 25 (23.4) | 35 (34.0) | 26 (25.7) | 17.7 (8.6, 26.8)c | 28.3 (18.2, 38.5)c | 0 | 27 (22.1) | 15 (12.2) | 22.1 (14.8, 29.5)c | 12.2 (6.4, 18.0)c |
| Minimal Disease Activity (MDA) n (%) | |||||||||||
| Week 24 | 16 (15.1) | 32 (29.9) | 42 (40.8) | 32 (31.7) | 14.8 (3.8, 25.8)a | 25.7 (14.0, 37.4)c | 4 (3.4) | 34 (27.9) | 29 (23.6) | 24.5 (15.9, 33.1)c | 20.2 (12.0, 28.4)c |
| ACR 50 and PASI 100 in patients with ≥3% BSA psoriasis skin involvement at baseline, n (%) | |||||||||||
| Week 24 | 1 (1.5) | 21 (28.8) | 19 (32.2) | 9 (13.2) | 27.3 (16.5, 38.1)c | 30.7 (18.4, 43.0)b | 0 (0.0) | 12 (17.6) | 10 (14.7) | 17.6 (8.6, 26.7)c | 14.7 (6.3, 23.1)c |
Abbreviations: ACR 20/50/70 = American College of Rheumatology 20%/50%/70% response rate; ADA = adalimumab; BSA = body surface area; CI = confidence interval; Q4W = ixekizumab 80 mg every 4 weeks; Q2W = ixekizumab 80 mg every 2 weeks; N = number of patients in the analysis population; n = number of patients in the specified category; NRI = non-responder imputation; PASI 100 = psoriasis area and severity index 100% improvement; PBO = placebo.
Note: patients who were rescued at week 16 or discontinued or with missing data were imputed as non-responders for week 24 analyses.
Concomitant cDMARDs included MTX, leflunomide and sulfasalazine.
a p<0.05; bp<0.01; cp<0.001 compared with placebo.
In patients with pre-existing dactylitis or enthesitis, treatment with ixekizumab Q4W resulted in improvement in dactylitis and enthesitis at week 24 compared to placebo (resolution: 78% vs. 24%; p<0.001, and 39% vs. 21%; p<0.01, respectively).
In patients with ≥3% BSA, the improvement in skin clearance at week 12 as measured by 75% improvement in Psoriasis Area Severity Index (PASI 75), was 67% (94/141) for those treated with the Q4W dosing regimen, and 9% (12/134) for those treated with placebo (p<0.001). The proportion of patients achieving a PASI 75, PASI 90, and PASI 100 response at week 24 was greater with ixekizumab Q4W compared to placebo (p<0.001). In patients with concomitant moderate to severe psoriasis and psoriatic arthritis, ixekizumab Q2W dose regimen showed significantly higher response rate for PASI75, PASI 90 and PASI 100 compared to placebo (p<0.001) and demonstrated clinically meaningful benefit over the Q4W dose regimen.
Treatment responses on ixekizumab were significantly greater than those on placebo as early as week 1 for ACR 20, week 4 for ACR 50 and week 8 for ACR 70 and persisted through week 24; effects were maintained through 3 years for patients who remained in the study.
Figure 4. ACR 20 response in SPIRIT-P1 over time up to week 24:

For both ixekizumab Q2W and Q4W: bp<0.01 and cp<0.001 compared with placebo.
In SPIRIT-P1 and SPIRIT-P2, similar responses for ACR 20/50/70 were seen in patients with psoriatic arthritis regardless of whether they were on concomitant cDMARDs, including MTX treatment, or not.
In SPIRIT-P1 and SPIRIT-P2, improvements were shown in all components of the ACR scores including patient assessment of pain. At week 24 the proportion of patients achieving a modified Psoriatic Arthritis Response Criteria (PsARC) response was greater in the ixekizumab-treated patients compared to placebo.
In SPIRIT-P1, efficacy was maintained up to Week 52 as assessed by ACR 20/50/70, MDA, enthesitis resolution, dactylitis resolution, and PASI 75/90/100 response rates.
The efficacy and safety of ixekizumab was demonstrated regardless of age, gender, race, disease duration, baseline body weight, baseline psoriasis involvement, baseline CRP, baseline DAS28-CRP, 51 concomitant corticosteroid use, and previous treatment with a biologic. Ixekizumab was efficacious in biologic-naive, biologic-exposed and biologic-failure patients.
In SPIRIT-P1, 63 patients completed 3 years of Q4W ixekizumab treatment. Among the 107 patients who were randomised to ixekizumab Q4W (NRI analysis in ITT population), 54 patients (50%), 41 patients (38%), 29 patients (27%), and 36 patients (34%) were observed to have ACR20, ACR50, ACR70, and MDA response, respectively, at week 156.
In SPIRIT-P2, 70 patients completed 3 years of Q4W ixekizumab treatment. Among the 122 patients who were randomised to ixekizumab Q4W (NRI analysis in ITT population), 56 patients (46%), 39 patients (32%), 24 patients (20%) and 33 (27%) were observed to have ACR20, ACR50, ACR70, and MDA response, respectively, at week 156.
Radiographic response
In SPIRIT-P1, inhibition of progression of structural damage was assessed radiographically and expressed as the change in modified total Sharp Score (mTSS) and its components, the Erosion Score (ES) and the Joint Space Narrowing score (JSN) at weeks 24 and 52, compared to baseline. week 24 data are presented in Table 11.
Table 11. Change in modified Total Sharp Score in SPIRIT-P1:
| Difference from placebo (95% CI) | ||||||
|---|---|---|---|---|---|---|
| PBO (N=106) | Ixekizumab Q4W (N=107) | Ixekizumab Q2W (N=103) | ADA (N=101) | Ixekizumab Q4W | Ixekizumab Q2W | |
| Baseline score, mean (SD) | 17.6 (28.62) | 19.2 (32.68) | 15.2 (28.86) | 15.9 (27.37) | NA | NA |
| Change from baseline at week 24, LSM (SE) | 0.51 (0.092) | 0.18 (0.090) | 0.09 (0.091) | 0.13 (0.093) | -0.33 (-0.57, -0.09)b | -0.42 (-0.66, -0.19)c |
Abbreviations: ADA = adalimumab; CI = confidence interval; Q4W = ixekizumab 80 mg every 4 weeks; Q2W = ixekizumab 80 mg every 2 weeks; LSM = least squares mean; N = number of patients in the analysis population; PBO = placebo; SE = standard error; SD = standard deviation.
b p<0.01; cp<0.001 compared with placebo.
Radiographic joint damage progression was inhibited by ixekizumab (Table 11) at week 24, and the percentage of patients with no radiographic joint damage progression (defined as a change from baseline in mTSS of ≤0.5) from randomisation to week 24 was 94.8% for ixekizumab Q2W (p<0.001), 89.0% for ixekizumab Q4W (p=0.026), 95.8% for adalimumab (p<0.001), all compared to 77.4% for placebo. At week 52, the mean change from baseline in mTSS was 0.27 for placebo/ixekizumab Q4W, 0.54 for ixekizumab Q4W/ixekizumab Q4W, and 0.32 for adalimumab/ixekizumab Q4W. The percentage of patients with no radiographic joint damage progression from randomisation to week 52 was 90.9% for placebo/ixekizumab Q4W, 85.6% for ixekizumab Q4W/ixekizumab Q4W, and 89.4% for adalimumab/ixekizumab Q4W. Patients had no structural progression from baseline (defined as mTSS≤0.5) in the treatment arms as follows: Placebo/ixekizumab Q4W 81.5% (N=22/27), ixekizumab Q4W/ixekizumab Q4W 73.6% (N=53/72), and adalimumab/ixekizumab Q4W 88.2% (N=30/34).
Physical function and health-related quality of life
In both SPIRIT-P1 and SPIRIT-P2, patients treated with Taltz Q2W (p<0.001) and Q4W (p<0.001) showed significant improvement in physical function compared to patients treated with placebo as assessed by Health Assessment Questionnaire-Disability Index (HAQ-DI) at Week 24, and maintained at Week 52 in SPIRIT-P1.
Ixekizumab-treated patients reported improvements in health-related quality of life as measured by the Physical Component Summary of the Short Form-36 Health Survey (SF-36 PCS) score (p<0.001). There were also improvements demonstrated in fatigue as assessed by Fatigue severity NRS scores (p<0.001).
Post-marketing phase 4, direct comparative study
Efficacy and safety of ixekizumab was investigated in a multicenter, randomised, open-label, rater-blinded, parallel-group study (SPIRIT-H2H) compared to adalimumab (ADA) in 566 patients with PsA who were naïve to biologic disease-modifying anti-rheumatic drugs (bDMARD). Patients were stratified at baseline based on concomitant cDMARD use and presence of moderate-to-severe psoriasis (PASI≥12, BSA≥10 and sPGA≥3).
Ixekizumab was superior to ADA on the primary study objective: simultaneous achievement of ACR 50 and PASI 100 response at week 24 (ixekizumab 36.0% vs ADA 27.9%; p=0.036; 95% confidence interval [0.5%, 15.8%]). Ixekizumab also showed non-inferiority (pre-specified margin of -12%) to ADA on ACR 50 (ITT analysis: ixekizumab 50.5% vs ADA 46.6%; 3.9% difference vs. ADA; 95% confidence interval [-4.3%; 12.1%]; PPS analysis ixekizumab: 52.3%, ADA: 53.1%, difference: -0.8% [CI: -10.3%; 8.7%]) and superiority on PASI 100 at week 24 (60.1% with ixekizumab vs 46.6% with ADA, p=0.001), which were the major secondary endpoints in the study.
At week 52 a higher proportion of patients treated with ixekizumab versus ADA simultaneously achieved ACR50 and PASI 100 [39% (111/283) versus 26% (74/283)] and PASI 100 [64% (182/283) versus 41% (117/283)]. Ixekizumab and ADA treatment resulted in similar responses for ACR50 [49.8% (141/283) versus 49.8% (141/283)]. Responses to ixekizumab were consistent when used as monotherapy or with concomitant use of methotrexate.
Figure 5. Primary endpoint (simultaneous ACR 50 & PASI 100) and major secondary endpoints (ACR 50; PASI 100) response rates week 0 – 24 [ITT population, NRI]**:
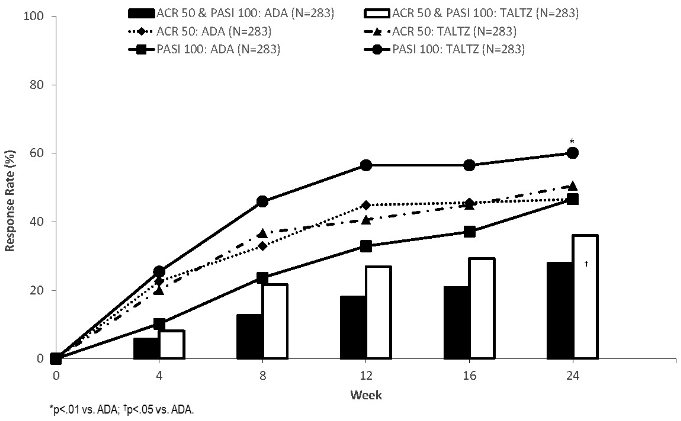
** Ixekizumab 160 mg week 0, then 80 mg every 2 weeks to week 12 and every 4 weeks thereafter for patients with moderate to severe plaque psoriasis or 160 mg week 0, then 80 mg every 4 week for other patients, ADA 80 mg week 0, then 40 mg every 2 weeks from week 1 for patients with moderate to severe plaque psoriasis or 40 mg week 0, then 40 mg every 2 weeks for other patients. Significance level only provided for endpoint that was pre-defined and multiplicity tested.
Axial spondyloarthritis
Ixekizumab was assessed in a total of 960 adult patients with axial spondyloarthritis in three randomised placebo-controlled studies (two in radiographic and one in non-radiographic axial spondyloarthritis).
Radiographic axial spondyloarthritis
Ixekizumab was assessed in a total of 657 patients in two randomised, double-blind, placebo-controlled studies (COAST-V and COAST-W) in adult patients who had active disease as defined by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 and total back pain ≥4 on a numeric rating scale despite non-steroidal anti-inflammatory drug (NSAID) therapy. Across both studies at baseline, patients had symptoms for a mean of 17 years (median of 16 years). At baseline, approximately 32% of the patients were on a concomitant cDMARD.
COAST-V evaluated 341 biologic-naive patients treated with either ixekizumab 80 mg or 160 mg at week 0 followed by 80 mg every 2 weeks (Q2W) or 4 weeks (Q4W), adalimumab 40 mg every 2 weeks, or with placebo. Patients receiving placebo were re-randomised at week 16 to receive ixekizumab (160 mg starting dose, followed by 80 mg Q2W or Q4W). Patients receiving adalimumab were re-randomised at week 16 to receive ixekizumab (80 mg Q2W or Q4W).
COAST-W evaluated 316 patients who had prior experience with 1 or 2 TNF-inhibitors (90% were inadequate responders and 10% were intolerant to TNF inhibitors). All patients were treated with ixekizumab 80 or 160 mg at week 0 followed by 80 mg Q2W or Q4W, or with placebo. Patients receiving placebo were re-randomised at week 16 to receive ixekizumab (160 mg initial dose, followed by 80 mg Q2W or Q4W).
The primary endpoint in both studies was the percentage of patients achieving an Assessment of Spondyloarthritis International Society 40 (ASAS40) response at week 16.
Clinical response
In both studies, patients treated with ixekizumab 80 mg Q2W or 80 mg Q4W demonstrated greater improvements in ASAS40 and ASAS20 responses compared to placebo at week 16 (Table 12). Responses were similar in patients regardless of concomitant therapies. In COAST-W, responses were seen regardless of the number of prior TNF inhibitors.
Table 12. Efficacy results in COAST-V and COAST-W at week 16:
| COAST-V, biologic-naive | COAST-W, TNF-inhibitor experienced | ||||||
|---|---|---|---|---|---|---|---|
| Ixekizumab 80 mg Q4Wa (N=81) | Placebo (N=87) | Difference from placebog | Adalimumab 40 mg Q2W (N=90) | Ixekizumab 80 mg Q4Wc (N=114) | Placebo (N=104) | Difference from placebog | |
| ASAS20 responseb, n (%), NRI | 52 (64.2%) | 35 (40.2%) | 24.0 (9.3, 38.6)** | 53 (58.9%) | 55 (48.2%) | 31 (29.8%) | 18.4 (5.7, 31.1)** |
| ASAS40 responseb,c, n (%), NRI | 39 (48.1%) | 16 (18.4%) | 29.8 (16.2, 43.3)*** | 32 (35.6%) | 29 (25.4%) | 13 (12.5%) | 12.9 (2.7, 23.2)* |
| ASDAS | |||||||
| Change from baseline Baseline | -1.4 3.7 | -0.5 3.9 | -1.0 (-1.3, -0.7)*** | -1.3*** 3.7 | -1.2 4.2 | -0.1 4.1 | -1.1 (-1.3, -0.8)*** |
| BASDAI Score | |||||||
| Change from baseline Baseline | -2.9 6.8i | -1.4 6.8i | -1.5 (-2.1, -0.9)*** | -2.5*** 6.7i | -2.2 7.5 | -0.9 7.3 | -1.2 (-1.8, -0.7)*** |
| MRI Spine SPARCCd | |||||||
| Change from baseline Baseline | -11.0 14.5 | -1.5 15.8 | -9.5 (-12.6, -6.4)*** | -11.6*** 20.0 | -3.0 8.3 | 3.3 6.4 | -6.3 (-10.0, -2.5)** |
| BASDAI50e n (%), NRI | 34 (42.0%) | 15 (17.2%) | 24.7 (11.4, 38.1)*** | 29 (32.2%)* | 25 (21.9%)i | 10 (9.6%)i | 12.3 (2.8, 21.8)* |
| ASDAS <2.1, n (%) (low disease activity), NRI | 35 (43.2%)h | 11 (12.6%)h | 30.6 (17.7,43.4)*** | 34 (37.8%)***h | 20 (17.5%) | 5 (4.8%) | 12.7 (4.6, 20.8)** |
| ASDAS <1.3, n (%) (inactive disease), NRI | 13 (16.0%) | 2 (2.3%) | 13.8 (5.2, 22.3)** | 14 (15.6%)** | 4 (3.5%)i | 1 (1.0%)i | 2.5 (-1.3, 6.4) |
| ASAS HIf Change from baseline Baseline | -2.4 7.5 | -1.3 8.1 | -1.1 (-2.0, -0.3)* | -2.3* 8.2 | -1.9 10.0 | -0.9 9.0 | -1.0 (-1.9, -0.1)* |
| SF-36 PCS Change from baseline Baseline | 7.7 34.0 | 3.6 32.0 | 4.1 (1.9, 6.2)*** | 6.9** 33.5 | 6.6 27.5 | 1.4 30.6 | 5.2 (3.0, 7.4)*** |
Abbreviations: N = number of patients in the intent-to-treat population; NRI = Non-responder Imputation; patients with missing data were counted as non-responders. ASAS HI = Assessment of SpondyloArthritis International Society Health Index; ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CFB = least square mean change from baseline at week 16; MRI Spine SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the Spine (23 discovertebral unit scale)
a At week 0, patients received 80 mg or 160 mg of ixekizumab.
b An ASAS20 response is defined as a ≥20% improvement and an absolute improvement from baseline of ≥1 unit (range 0 to 10) in ≥3 of 4 domains (Patient Global, Spinal Pain, Function, and Inflammation), and no worsening of ≥20% and ≥1 unit (range 0 to 10) in the remaining domain. An ASAS40 response is defined as a ≥40% improvement and an absolute improvement from baseline of ≥2 units in ≥3 of 4 domains without any worsening in the remaining domain.
c Primary endpoint.
d The numbers of ITT patients with MRI data at baseline are as follows: COAST-V: ixekizumab, n=81; PBO, n=82; ADA, n=85. COAST-W: ixekizumab, n=58; PBO, n=51.
e BASDAI50 response defined as an improvement of ≥50% of the BASDAI score from baseline.
f ASAS HI: Assessment of SpondyloArthritis International Society Health Index (ASAS HI) across all domains.
g The reported values are difference in % (95% CI) for categorical variables, and difference in LSM (95% CI) for continuous variables.
h post hoc analysis, not multiplicity corrected.
i prespecified, but not multiplicity gated.
* p<0.05; p<0.01; *p<0.001 compared with placebo.
There were improvements in the main components of the ASAS40 response criteria (spinal pain, BASFI, patient global assessment, stiffness) and other measures of disease activity, including CRP, at week 16.
Figure 6. Percent of patients achieving ASAS40 responses in COAST-V and COAST-W through week 16, NRIa:
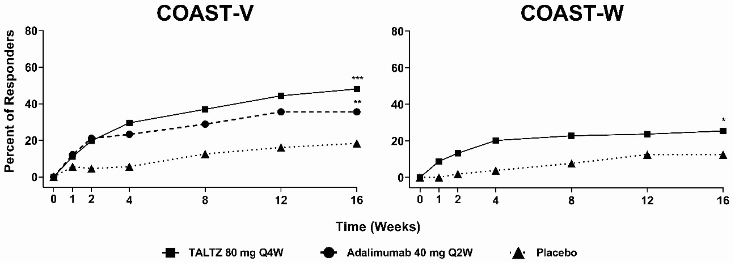
a Patients with missing data were counted as non-responders.
* p<0.05; p<0.01; *p<0.001 compared with placebo.
Similar response in ASAS40 was seen in patients regardless of baseline CRP levels, baseline ASDAS scores and MRI spine SPARCC scores. The ASAS40 response was demonstrated regardless of age, gender, race, disease duration, baseline body weight, baseline BASDAI score and prior biologic treatment.
In COAST-V and COAST-W efficacy was maintained up to week 52 as assessed by the endpoints presented in Table 12, including ASAS20, ASAS40, ASDAS, BASDAI, and ASAS HI response rates.
Health-related outcomes
Spinal pain showed improvements versus placebo as early as week 1, maintained through week 16 [ixekizumab vs placebo: COAST-V -3.2 vs -1.7; COAST-W -2.4 vs -1.0]; fatigue and spinal mobility showed improvements versus placebo at week 16. Improvements in spinal pain, fatigue and spinal mobility were maintained through week 52.
Non-radiographic axial spondyloarthritis
Ixekizumab was assessed in a randomised, double-blind study with a 52-week placebo-controlled period (COAST-X) in 303 adult patients with active axial spondyloarthritis for at least 3 months. Patients must have had objective signs of inflammation indicated by elevated C-reactive protein (CRP) and/or sacroiliitis on magnetic resonance imaging (MRI), and no definitive radiographic evidence of structural damage on sacroiliac joints. Patients had active disease as defined by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, and spinal pain ≥4 on a 0 to 10 Numerical Rating Scale (NRS), despite non-steroidal anti-inflammatory drug (NSAID) therapy. Patients were treated with either ixekizumab 80 mg or 160 mg at week 0, followed by 80 mg every 2 weeks (Q2W) or 80 mg every 4 weeks (Q4W) or with placebo. Dose adjustment and/or initiation of concomitant medications (NSAIDs, cDMARDs, corticosteroids, analgesics) were permitted starting at week 16.
At baseline, patients had symptoms of non-radiographic axSpA for an average of 11 years. Approximately 39% of the patients were on a concomitant cDMARD.
The primary endpoint was the percentage of patients achieving an Assessment of Spondyloarthritis International Society 40 (ASAS40) response at week 16.
Clinical response
Higher proportions of patients treated with ixekizumab 80 mg Q4W achieved ASAS40 response compared to placebo at week 16 (Table 13). Responses were similar regardless of concomitant therapies.
Table 13. Efficacy results at week 16 in COAST-X, NRIa,b:
| Ixekizumab 80 mg Q4Wc (N=96) | Placebo (N=105) | Difference from placeboh | |
|---|---|---|---|
| ASAS20 responsed, n (%), NRI | 52 (54.2%) | 41 (39.0%) | 15.1 (1.5, 28.8)* |
| ASAS40 responsed,e, n (%), NRI | 34 (35.4%) | 20 (19.0%) | 16.4 (4.2, 28.5)** |
| ASDAS | |||
| Change from baseline Baseline | -1.1 3.8 | -0.6 3.8 | -0.5 (-0.8, -0.3)*** |
| BASDAI Score | |||
| Change from baseline Baseline | -2.2 7.0 | -1.5 7.2 | -0.7 (-1.3, -0.1)* |
| MRI SIJ SPARCCf | |||
| Change from baseline Baseline | -3.4 5.1 | -0.3 6.3 | -3.1 (-4.6, -1.6)*** |
| ASDAS <2.1, n (%) (low disease activity), NRIg | 26 (27.7%) | 13 (12.4%) | 15.3 (4.3, 26.3)** |
| SF-36 PCS | |||
| Change from baseline Baseline | 8.1 33.5 | 5.2 32.6 | 2.9 (0.6, 5.1)* |
a Abbreviations: N = number of patients in the intent-to-treat population; NRI = Non-responder Imputation. ASDAS = Ankylosing Spondylitis Disease Activity Score; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; Change from baseline = least square mean change from baseline at week 16; MRI SIJ SPARCC = Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Scoring of the sacroiliac joint.
b Patients with missing data were counted as non-responders.
c At week 0, patients received 80 mg or 160 mg of ixekizumab.
d An ASAS20 response is defined as a ≥20% improvement and an absolute improvement from baseline of ≥1 units (range 0 to 10) in ≥3 of 4 domains (Patient Global, Spinal Pain, Function, and Inflammation), and no worsening of ≥20% and ≥1 unit (range 0 to 10) in the remaining domain. An ASAS40 response is defined as a ≥40% improvement and an absolute improvement from baseline of ≥2 units in ≥3 of 4 domains without any worsening in the remaining domain.
e Primary endpoint at week 16.
f The numbers of ITT patients with MRI data at baseline and week 16 are as follows: ixekizumab, n=85; PBO, n=90.
g Patients with missing data were counted as non-responders. Percentages are based on the number of patients in the ITT population with baseline ASDAS ≥2.1.
h The reported values are difference in ( 95 CI) for categorical variables, and difference in LSM (95% CI) for continuous variables.
* p<0.05; p<0.01; *p<0.001 compared with placebo.
The improvement in the main components of the ASAS40 response criteria (spinal pain, BASFI, patient global assessment, stiffness) and other measures of disease activity demonstrated significant clinical improvement at week 16.
Figure 7. Percent of patients achieving ASAS40 response through week 16 in COAST-X, NRIa:
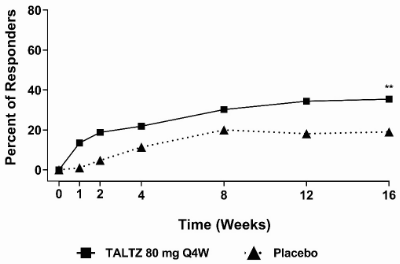
a Patients with missing data were counted as non-responders.
** p<0.01 compared with placebo.
Efficacy was maintained up to week 52 as assessed by the endpoints presented in Table 13.
Health-related outcomes
Spinal pain showed improvements versus placebo as early as week 1 and was maintained through week 16 [ixekizumab vs placebo: COAST-X: -2.4 vs -1.5]. In addition, more patients on ixekizumab compared with placebo achieved good health status (ASAS HI ≤5) at week 16 and week 52.
Long-term outcomes Axial Spondyloarthritis
Patients who completed one of the three pivotal studies COAST-V/W/X (52 weeks) were offered participation in a long-term extension and randomised withdrawal study (COAST-Y, with 350 and 423 patients enrolled on ixekizumab Q4W and Q2W, respectively). Among those who achieved remission 157/773 (20.3%) (Ankylosing Spondylitis Disease Activity Score [ASDAS] <1.3 at least once, and no ASDAS score ≥2.1, at weeks 16 and 20), 155 patients exposed to ixekizumab up to 76 weeks were randomised at week 24 of the COAST-Y study (Placebo, N=53; ixekizumab Q4W, N=48; and ixekizumab Q2W, N=54); of these, 148 (95.5%) completed the week 64 visit (Placebo, N=50; ixekizumab Q4W, N=47; ixekizumab Q2W, N=51). The primary endpoint was the proportion of patients in the randomised withdrawal population who did not experience a flare during weeks 24-64 (combined ixekizumab Q2W and ixekizumab Q4W groups versus placebo). A significantly larger proportion of patients (NRI) in the combined ixekizumab groups (83.3% (85/102), p<0.001) and ixekizumab Q4W (83.3% (40/48), p=0.003) had no flare during weeks 24-64 compared with those who withdrew from ixekizumab to placebo (54.7% (29/53)). Ixekizumab (in both combined ixekizumab groups and ixekizumab Q4W group) significantly delayed the time to flare (Log-Rank Test p<0.001 and p<0.01, respectively) compared to Placebo.
In patients who received ixekizumab Q4W continuously (N=157), the ASAS40, ASDAS <2.1 and BASDAI50 responses were maintained to week 116.
Immunisations
In a study in healthy subjects, no safety concerns were identified of two inactivated vaccines (tetanus and pneumococcal), received after two doses of ixekizumab (160 mg followed by a second dose of 80 mg two weeks later). However, the data concerning immunisation were insufficient to conclude on an adequate immune response to these vaccines following administration of ixekizumab.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with ixekizumab in one or more subsets of the paediatric population in the treatment of plaque psoriasis and psoriatic arthritis/axial spondyloarthritis (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
Following a single subcutaneous dose of ixekizumab in patients with psoriasis, mean peak concentrations were achieved within 4 to 7 days, across a dose range of 5 to 160 mg. The mean (SD) maximum plasma concentration (Cmax) of ixekizumab, after the 160 mg starting dose, was 19.9 (8.15) µg/ml.
After the 160 mg starting dose, steady state was achieved by Week 8 with the 80 mg Q2W dosing regimen. Mean (SD) Cmax,ss, and Ctrough,ss estimates are 21.5 (9.16) µg/ml, and 5.23 (3.19) µg/ml.
After switching from the 80 mg Q2W dosing regimen to the 80 mg Q4W dosing regimen at Week 12, steady state would be achieved after approximately 10 weeks. Mean (SD) Cmax,ss, and Ctrough,ss estimates are 14.6 (6.04) µg/ml, and 1.87 (1.30) µg/ml.
The average bioavailability of ixekizumab after subcutaneous administration was 54% to 90% across analyses.
Distribution
From population pharmacokinetic analyses, the mean total volume of distribution at steady state was 7.11 L.
Biotransformation
Ixekizumab is a monoclonal antibody and is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous immunoglobulins.
Elimination
In the population PK analysis, mean serum clearance was 0.0161 L/hr. Clearance is independent of dose. The mean elimination half-life, as estimated from population pharmacokinetic analysis, is 13 days in patients with plaque psoriasis.
Linearity/non-linearity
Exposure (Area Under the Curve or AUC) increased proportionally over a dose range of 5 to 160 mg given as a subcutaneous injection.
Pharmacokinetic properties across indications
The pharmacokinetic properties of ixekizumab were similar across the plaque psoriasis, psoriatic arthritis, radiographic axial spondyloarthritis and non-radiographic axial spondyloarthritis indications.
Elderly
Of the 4,204 plaque psoriasis patients exposed to ixekizumab in clinical studies, a total of 301 were 65 years of age or older and 36 patients were 75 years of age or older. Of the 1 118 psoriatic arthritis patients exposed to ixekizumab in clinical studies, a total of 122 patients were 65 years of age or older and 6 patients were 75 years of age or older. Based on population pharmacokinetic analysis with a limited number of elderly patients (n=94 for age ≥65 years and n=12 for age ≥75 years), clearance in elderly patients and patients less than 65 years of age was similar.
Renal or hepatic impairment
Specific clinical pharmacology studies to evaluate the effects of renal impairment and hepatic impairment on the PK of ixekizumab have not been conducted. Renal elimination of intact ixekizumab, an IgG MAb, is expected to be low and of minor importance; similarly, IgG MAbs are mainly eliminated via intracellular catabolism and hepatic impairment is not expected to influence clearance of ixekizumab.
Paediatric population
Paediatric psoriasis patients (age 6 to less than 18 years) were administered ixekizumab at the recommended paediatric dosing regimen for 12 weeks. Patients weighing >50 kg and 25 to 50 kg had a mean ±SD steady-state trough concentration of 3.8 ± 2.2 μg/mL and 3.9 ± 2.4 μg/mL, respectively, at week 12.
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on repeat-dose toxicity studies, safety pharmacology evaluations, and reproductive and developmental toxicity studies.
Ixekizumab administration to cynomolgus monkeys for 39 weeks at subcutaneous doses up to 50 mg/kg weekly produced no organ toxicity or undesirable effects on immune function (e.g. T-cell dependent antibody response and NK cell activity). A weekly subcutaneous dose of 50 mg/kg to monkeys is approximately 19 times the 160 mg starting dose of ixekizumab and in monkeys results in exposure (AUC) that is at least 61-fold higher than the predicted mean steady-state exposure in humans administered the recommended dose regimen.
Non-clinical studies have not been conducted to evaluate the carcinogenic or mutagenic potential of ixekizumab.
No effects on reproductive organs, menstrual cycles or sperm were observed in sexually mature cynomolgus monkeys that received ixekizumab for 13 weeks at a weekly subcutaneous dose of 50 mg/kg.
In developmental toxicity studies, ixekizumab was shown to cross the placenta and was present in the blood of offspring for up to 6 months of age. A higher incidence of postnatal mortality occurred in the offspring of monkeys given ixekizumab compared to concurrent controls. This was related primarily to early delivery or maternal neglect of offspring, common findings in nonhuman primate studies, and considered clinically irrelevant.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.