TARCEVA Film-coated tablet Ref.[8171] Active ingredients: Erlotinib
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Roche Registration GmbH, Emil-Barell-Strasse 1, 79639, Grenzach-Wyhlen, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agent protein kinase inhibitor
ATC code: L01XE03
Mechanism of action
Erlotinib is an epidermal growth factor receptor/human epidermal growth factor receptor type 1 (EGFR also known as HER1) tyrosine kinase inhibitor. Erlotinib potently inhibits the intracellular phosphorylation of EGFR. EGFR is expressed on the cell surface of normal cells and cancer cells. In non-clinical models, inhibition of EGFR phosphotyrosine results in cell stasis and/or death.
EGFR mutations may lead to constitutive activation of anti-apoptotic and proliferation signaling pathways. The potent effectiveness of erlotinib in blocking EGFR-mediated signalling in these EGFR mutation positive tumours is attributed to the tight binding of erlotinib to the ATP-binding site in the mutated kinase domain of the EGFR. Due to the blocking of downstream-signaling, the proliferation of cells is stopped, and cell death is induced through the intrinsic apoptotic pathway. Tumour regression is observed in mouse models of enforced expression of these EGFR activating mutations.
Clinical efficacy
First-line Non-Small Cell Lung Cancer (NSCLC) therapy for patients with EGFR activating mutations (Tarceva administered as monotherapy)
The efficacy of Tarceva in first-line treatment of patients with EGFR activating mutations in NSCLC was demonstrated in a phase III, randomised, open-label trial (ML20650, EURTAC). This study was conducted in Caucasian patients with metastatic or locally advanced NSCLC (stage IIIB and IV) who have not received previous chemotherapy or any systemic antitumour therapy for their advanced disease and who present mutations in the tyrosine kinase domain of the EGFR (exon 19 deletion or exon 21 mutation). Patients were randomised 1:1 to receive Tarceva 150 mg daily or up to 4 cycles of platinum based doublet chemotherapy.
The primary endpoint was investigator assessed PFS. The efficacy results are summarized in Table 3.
Figure 1. Kaplan-Meier curve for investigator assessed PFS in trial ML20650 (EURTAC) (April 2012 cut-off):

Table 3. Efficacy results of Tarceva versus chemotherapy in trial ML20650 (EURTAC):
Maintenance NSCLC therapy after first-line chemotherapy (Tarceva administered as monotherapy)
The efficacy and safety of Tarceva as maintenance after first-line chemotherapy for NSCLC was investigated in a randomised, double-blind, placebo-controlled trial (BO18192, SATURN). This study was conducted in 889 patients with locally advanced or metastatic NSCLC who did not progress after 4 cycles of platinum-based doublet chemotherapy. Patients were randomised 1:1 to receive Tarceva 150 mg or placebo orally once daily until disease progression. The primary endpoint of the study included progression free survival (PFS) in all patients. Baseline demographic and disease characteristics were well balanced between the two treatment arms. Patients with ECOG PS>1, significant hepatic or renal co-morbidities were not included in the study.
In this study, the overall population showed a benefit for the primary PFS end-point (HR= 0.71 p<0.0001) and the secondary OS end-point (HR=0.81 p=0.0088). However the largest benefit was observed in a predefined exploratory analysis in patients with EGFR activating mutations (n=49) demonstrating a substantial PFS benefit (HR=0.10, 95% CI, 0.04 to 0.25; p<0.0001) and an overall survival HR of 0.83 (95% CI, 0.34 to 2.02). 67% of placebo patients in the EGFR mutation positive subgroup received second or further line treatment with EGFR-TKIs.
The BO25460 (IUNO) study was conducted in 643 patients with advanced NSCLC whose tumors did not harbor an EGFR-activating mutation (exon 19 deletion or exon 21 L858R mutation) and who had not experienced disease progression after four cycles of platinum-based chemotherapy.
The objective of the study was to compare the overall survival of first line maintenance therapy with erlotinib versus erlotinib administered at the time of disease progression. The study did not meet its primary endpoint. OS of Tarceva in first line maintenance was not superior to Tarceva as second line treatment in patients whose tumor did not harbor an EGFR-activating mutation (HR=1.02, 95% CI, 0.85 to 1.22, p=0.82). The secondary endpoint of PFS showed no difference between Tarceva and placebo in maintenance treatment (HR=0.94, 95% CI, 0.80 to 1.11; p=0.48).
Based on the data from the BO25460 (IUNO) study, Tarceva use is not recommended for first-line maintenance treatment in patients without an EGFR activating mutation.
NSCLC treatment after failure of at least one prior chemotherapy regimen (Tarceva administered as monotherapy)
The efficacy and safety of Tarceva as second/third-line therapy was demonstrated in a randomised, double-blind, placebo-controlled trial (BR.21), in 731 patients with locally advanced or metastatic NSCLC after failure of at least one chemotherapy regimen. Patients were randomised 2:1 to receive Tarceva 150 mg or placebo orally once daily. Study endpoints included overall survival, progression-free survival (PFS), response rate, duration of response, time to deterioration of lung cancer-related symptoms (cough, dyspnoea and pain), and safety. The primary endpoint was survival.
Demographic characteristics were well balanced between the two treatment groups. About two-thirds of the patients were male and approximately one-third had a baseline ECOG performance status (PS) of 2, and 9% had a baseline ECOG PS of 3. Ninety-three percent and 92% of all patients in the Tarceva and placebo groups, respectively, had received a prior platinum-containing regimen and 36% and 37% of all patients, respectively, had received a prior taxane therapy.
The adjusted hazard ratio (HR) for death in the Tarceva group relative to the placebo group was 0.73 (95% CI, 0.60 to 0.87) (p=0.001). The percent of patients alive at 12 months was 31.2% and 21.5%, for the Tarceva and placebo groups, respectively. The median overall survival was 6.7 months in the Tarceva group (95% CI, 5.5 to 7.8 months) compared with 4.7 months in the placebo group (95% CI, 4.1 to 6.3 months).
The effect on overall survival was explored across different patient subsets. The effect of Tarceva on overall survival was similar in patients with a baseline performance status (ECOG) of 2-3 (HR=0.77, 95% CI 0.6-1.0) or 0-1 (HR=0.73, 95% CI 0.6-0.9), male (HR=0.76, 95% CI 0.6-0.9) or female patients (HR=0.80, 95% CI 0.6-1.1), patients < 65 years of age (HR=0.75, 95% CI 0.6-0.9) or older patients (HR=0.79, 95% CI 0.6-1.0), patients with one prior regimen (HR=0.76, 95% CI 0.6-1.0) or more than one prior regimen (HR=0.75, 95% CI 0.6-1.0), Caucasian (HR=0.79, 95% CI 0.6-1.0) or Asian patients (HR=0.61, 95% CI 0.4-1.0), patients with adenocarcinoma (HR=0.71, 95% CI 0.6-0.9) or squamous cell carcinoma (HR=0.67, 95% CI 0.5-0.9), but not in patients with other histologies (HR 1.04, 95% CI 0.7-1.5), patients with stage IV disease at diagnosis (HR=0.92, 95% CI 0.7-1.2) or < stage IV disease at diagnosis (HR=0.65, 95% CI 0.5-0.8). Patients who never smoked had a much greater benefit from erlotinib (survival HR=0.42, 95% CI 0.28-0.64) compared with current or ex-smokers (HR=0.87, 95% CI 0.71-1.05).
In the 45% of patients with known EGFR-expression status, the hazard ratio was 0.68 (95% CI 0.49-0.94) for patients with EGFR-positive tumours and 0.93 (95% CI 0.63-1.36) for patients with EGFR-negative tumours (defined by IHC using EGFR pharmDx kit and defining EGFR-negative as less than 10% tumour cells staining). In the remaining 55% of patients with unknown EGFR-expression status, the hazard ratio was 0.77 (95% CI 0.61-0.98).
The median PFS was 9.7 weeks in the Tarceva group (95% CI, 8.4 to 12.4 weeks) compared with 8.0 weeks in the placebo group (95% CI, 7.9 to 8.1 weeks).
The objective response rate by RECIST in the Tarceva group was 8.9% (95% CI, 6.4 to 12.0). The first 330 patients were centrally assessed (response rate 6.2%); 401 patients were investigator- assessed (response rate 11.2%).
The median duration of response was 34.3 weeks, ranging from 9.7 to 57.6+ weeks. The proportion of patients who experienced complete response, partial response or stable disease was 44.0% and 27.5%, respectively, for the Tarceva and placebo groups (p=0.004).
A survival benefit of Tarceva was also observed in patients who did not achieve an objective tumour response (by RECIST). This was evidenced by a hazard ratio for death of 0.82 (95% CI, 0.68 to 0.99) among patients whose best response was stable disease or progressive disease.
Tarceva resulted in symptom benefits by significantly prolonging time to deterioration in cough, dyspnoea and pain, versus placebo.
In a double-blind, randomised phase III study (MO22162, CURRENTS) comparing two doses of Tarceva (300 mg versus 150 mg) in current smokers (mean of 38 pack years) with locally advanced or metastatic NSCLC in the second-line setting after failure on chemotherapy, the 300 mg dose of Tarceva demonstrated no PFS benefit over the recommended dose (7.00 vs 6.86 weeks, respectively).
Secondary efficacy endpoints were all consistent with the primary endpoint and no difference was detected for OS between patients treated with erlotinib 300 mg and 150 mg daily (HR 1.03, 95% CI 0.80 to 1.32). Safety data were comparable between the 300 mg and 150 mg doses; however, there was a numerical increase in the incidence of rash, interstitial lung disease and diarrhoea, in patients receiving the higher dose of erlotinib. Based on the data from the CURRENTS study, no evidence was seen for any benefit of a higher erlotinib dose of 300 mg when compared with the recommended dose of 150 mg in active smokers.
Patients in this study were not selected based on EGFR mutation status. See sections 4.2, 4.4, 4.5, and 5.2.
Pancreatic cancer (Tarceva administered concurrently with gemcitabine in study PA.3)
The efficacy and safety of Tarceva in combination with gemcitabine as a first-line treatment was assessed in a randomised, double-blind, placebo-controlled trial in patients with locally advanced, unresectable or metastatic pancreatic cancer. Patients were randomised to receive Tarceva or placebo once daily on a continuous schedule plus gemcitabine IV (1000 mg/m², Cycle 1 – Days 1, 8, 15, 22, 29, 36 and 43 of an 8 week cycle; Cycle 2 and subsequent cycles – Days 1, 8 and 15 of a 4 week cycle [approved dose and schedule for pancreatic cancer, see the gemcitabine SPC]). Tarceva or placebo was taken orally once daily until disease progression or unacceptable toxicity. The primary endpoint was overall survival.
Baseline demographic and disease characteristics of the patients were similar between the 2 treatment groups, 100 mg Tarceva plus gemcitabine or placebo plus gemcitabine, except for a slightly larger proportion of females in the erlotinib/gemcitabine arm compared with the placebo/gemcitabine arm:
| Baseline | Tarceva | Placebo |
|---|---|---|
| Females | 51% | 44% |
| Baseline ECOG performance status (PS) = 0 | 31% | 32% |
| Baseline ECOG performance status (PS) = 1 | 51% | 51% |
| Baseline ECOG performance status (PS) = 2 | 17% | 17% |
| Metastatic disease at baseline | 77% | 76% |
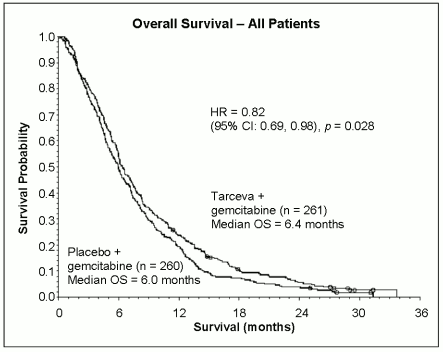
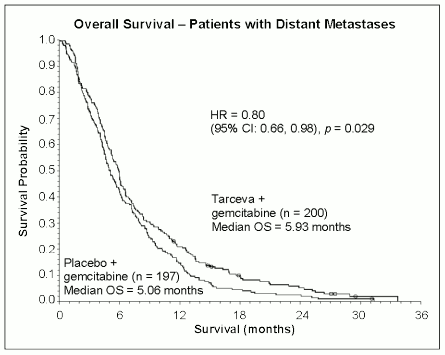
Survival was evaluated in the intent-to-treat population based on follow-up survival data. Results are shown in the table below (results for the group of metastatic and locally advanced patients are derived from exploratory subgroup analysis).
| Outcome | Tarceva (months) | Placebo (months) | ∆ (months) | CI of ∆ | HR | CI of HR | P-value |
|---|---|---|---|---|---|---|---|
| Overall Population | |||||||
| Median overall survival | 6.4 | 6.0 | 0.41 | -0.54-1.64 | 0.82 | 0.69-0.98 | 0.028 |
| Mean overall survival | 8.8 | 7.6 | 1.16 | -0.05-2.34 | |||
| Metastatic Population | |||||||
| Median overall survival | 5.9 | 5.1 | 0.87 | -0.26-1.56 | 0.80 | 0.66-0.98 | 0.029 |
| Mean overall survival | 8.1 | 6.7 | 1.43 0.17-2.66 | ||||
| Locally Advanced Population | |||||||
| Median overall survival | 8.5 | 8.2 | 0.36 | -2.43-2.96 | 0.93 | 0.65-1.35 | 0.713 |
| Mean overall survival | 10.7 | 10.5 | 0.19 | -2.43-2.69 | |||
In a post-hoc analysis, patients with favourable clinical status at baseline (low pain intensity, good QoL and good PS) may derive more benefit from Tarceva. The benefit is mostly driven by the presence of a low pain intensity score.
In a post-hoc analysis, patients on Tarceva who developed a rash had a longer overall survival compared to patients who did not develop rash (median OS 7.2 months vs 5 months, HR:0.61). 90% of patients on Tarceva developed rash within the first 44 days. The median time to onset of rash was 10 days.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Tarceva in all subsets of the paediatric population in Non Small Cell Lung Cancer and Pancreatic cancer indications (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
After oral administration, erlotinib peak plasma levels are obtained in approximately 4 hours after oral dosing. A study in normal healthy volunteers provided an estimate of the absolute bioavailability of 59%. The exposure after an oral dose may be increased by food.
Distribution
Erlotinib has a mean apparent volume of distribution of 232 l and distributes into tumour tissue of humans. In a study of 4 patients (3 with non-small cell lung cancer [NSCLC], and 1 with laryngeal cancer) receiving 150 mg daily oral doses of Tarceva, tumour samples from surgical excisions on Day 9 of treatment revealed tumour concentrations of erlotinib that averaged 1185 ng/g of tissue. This corresponded to an overall average of 63% (range 5-161%) of the steady state observed peak plasma concentrations. The primary active metabolites were present in tumour at concentrations averaging 160 ng/g tissue, which corresponded to an overall average of 113% (range 88-130%) of the observed steady state peak plasma concentrations. Plasma protein binding is approximately 95%. Erlotinib binds to serum albumin and alpha-1 acid glycoprotein (AAG).
Biotransformation
Erlotinib is metabolised in the liver by the hepatic cytochromes in humans, primarily CYP3A4 and to a lesser extent by CYP1A2. Extrahepatic metabolism by CYP3A4 in intestine, CYP1A1 in lung, and 1B1 in tumour tissue potentially contribute to the metabolic clearance of erlotinib.
There are three main metabolic pathways identified: 1) O-demethylation of either side chain or both, followed by oxidation to the carboxylic acids; 2) oxidation of the acetylene moiety followed by hydrolysis to the aryl carboxylic acid; and 3) aromatic hydroxylation of the phenyl-acetylene moiety. The primary metabolites OSI-420 and OSI-413 of erlotinib produced by O-demethylation of either side chain have comparable potency to erlotinib in non-clinical in vitro assays and in vivo tumour models. They are present in plasma at levels that are <10% of erlotinib and display similar pharmacokinetics as erlotinib.
Elimination
Erlotinib is excreted predominantly as metabolites via the faeces (>90%) with renal elimination accounting for only a small amount (approximately 9%) of an oral dose. Less than 2% of the orally administered dose is excreted as parent substance. A population pharmacokinetic analysis in 591 patients receiving single agent Tarceva shows a mean apparent clearance of 4.47 l/hour with a median half-life of 36.2 hours. Therefore, the time to reach steady state plasma concentration would be expected to occur in approximately 7-8 days.
Pharmacokinetics in special populations
Based on population pharmacokinetic analysis, no clinically significant relationship between predicted apparent clearance and patient age, bodyweight, gender and ethnicity were observed. Patient factors, which correlated with erlotinib pharmacokinetics, were serum total bilirubin, AAG and current smoking. Increased serum concentrations of total bilirubin and AAG concentrations were associated with a reduced erlotinib clearance. The clinical relevance of these differences is unclear. However, smokers had an increased rate of erlotinib clearance. This was confirmed in a pharmacokinetic study in non-smoking and currently cigarette smoking healthy subjects receiving a single oral dose of 150 mg erlotinib. The geometric mean of the Cmax was 1056 ng/mL in the non-smokers and 689 ng/mL in the smokers with a mean ratio for smokers to non-smokers of 65.2% (95% CI: 44.3 to 95.9, p=0.031). The geometric mean of the AUC0-inf was 18726 ng•h/mL in the non-smokers and 6718 ng•h/mL in the smokers with a mean ratio of 35.9% (95% CI: 23.7 to 54.3, p<0.0001). The geometric mean of the C24h was 288 ng/mL in the non-smokers and 34.8 ng/mL in the smokers with a mean ratio of 12.1% (95% CI: 4.82 to 30.2, p=0.0001). In the pivotal Phase III NSCLC trial, current smokers achieved erlotinib steady state trough plasma concentration of 0.65 μg/mL (n=16) which was approximately 2-fold less than the former smokers or patients who had never smoked (1.28 μg/mL, n=108). This effect was accompanied by a 24% increase in apparent erlotinib plasma clearance. In a phase I dose escalation study in NSCLC patients who were current smokers, pharmacokinetic analyses at steady-state indicated a dose proportional increase in erlotinib exposure when the Tarceva dose was increased from 150 mg to the maximum tolerated dose of 300 mg. Steady-state trough plasma concentrations at a 300 mg dose in current smokers in this study was 1.22 μg/mL (n=17). See sections 4.2, 4.4, 4.5 and 5.1.
Based on the results of pharmacokinetic studies, current smokers should be advised to stop smoking while taking Tarceva, as plasma concentrations could be reduced otherwise.
Based on population pharmacokinetic analysis, the presence of an opioid appeared to increase exposure by about 11%.
A second population pharmacokinetic analysis was conducted that incorporated erlotinib data from 204 pancreatic cancer patients who received erlotinib plus gemcitabine. This analysis demonstrated that covariants affecting erlotinib clearance in patients from the pancreatic study were very similar to those seen in the prior single agent pharmacokinetic analysis. No new covariate effects were identified. Co-administration of gemcitabine had no effect on erlotinib plasma clearance.
Paediatric population
There have been no specific studies in paediatric patients.
Elderly population
There have been no specific studies in elderly patients.
Hepatic impairment
Erlotinib is primarily cleared by the liver. In patients with solid tumours and with moderately impaired hepatic function (Child-Pugh score 7-9), geometric mean erlotinib AUC0-t and Cmax was 27000 ng•h/mL and 805 ng/mL, respectively, as compared to 29300 ng•h/mL and 1090 ng/mL in patients with adequate hepatic function including patients with primary liver cancer or hepatic metastases. Although the Cmax was statistically significant lower in moderately hepatic impaired patients, this difference is not considered clinically relevant. No data are available regarding the influence of severe hepatic dysfunction on the pharmacokinetics of erlotinib. In population pharmacokinetic analysis, increased serum concentrations of total bilirubin were associated with a slower rate of erlotinib clearance.
Renal impairment
Erlotinib and its metabolites are not significantly excreted by the kidney, as less than 9% of a single dose is excreted in the urine. In population pharmacokinetic analysis, no clinically significant relationship was observed between erlotinib clearance and creatinine clearance, but there are no data available for patients with creatinine clearance <15 ml/min.
Preclinical safety data
Chronic dosing effects observed in at least one animal species or study included effects on the cornea (atrophy, ulceration), skin (follicular degeneration and inflammation, redness, and alopecia), ovary (atrophy), liver (liver necrosis), kidney (renal papillary necrosis and tubular dilatation), and gastrointestinal tract (delayed gastric emptying and diarrhoea). Red blood cell parameters were decreased and white blood cells, primarily neutrophils, were increased. There were treatment-related increases in ALT, AST and bilirubin. These findings were observed at exposures well below clinically relevant exposures.
Based on the mode of action, erlotinib has the potential to be a teratogen. Data from reproductive toxicology tests in rats and rabbits at doses near the maximum tolerated dose and/or maternally toxic doses showed reproductive (embryotoxicity in rats, embryo resorption and foetotoxicity in rabbits) and developmental (decrease in pup growth and survival in rats) toxicity, but was not teratogenic and did not impair fertility. These findings were observed at clinically relevant exposures.
Erlotinib tested negative in conventional genotoxicity studies. Two-year carcinogenicity studies with erlotinib conducted in rats and mice were negative up to exposures exceeding human therapeutic exposure (up to 2-fold and 10-fold higher, respectively, based on Cmax and/or AUC).
A mild phototoxic skin reaction was observed in rats after UV irradiation.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.