TIBSOVO Film-coated tablet Ref.[109170] Active ingredients: Ivosidenib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Les Laboratoires Servier, 50, rue Carnot, 92284 Suresnes cedex, France
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents; other antineoplastic agents
ATC code: L01XX62
Mechanism of action
Ivosidenib is an inhibitor of the mutant IDH1 enzyme. Mutant IDH1 converts alpha- ketoglutarate (αKG) to 2-hydroxyglutarate (2-HG) which blocks cellular differentiation and promotes tumorigenesis in both hematologic and non-hematologic malignancies. The mechanism of action of ivosidenib beyond its ability to reduce 2-HG and restore cellular differentiation is not fully understood across indications.
Pharmacodynamic effects
Multiple doses of ivosidenib 500 mg daily decreased plasma concentrations of 2-HG in patients with hematological malignancies and cholangiocarcinoma with mutated IDH1 to levels approximating those observed in healthy subjects. In bone marrow of patients with hematological malignancies and in tumour biopsy of patients with cholangiocarcinoma, the mean (% coefficient of variation [CV]) reduction in 2-HG concentrations were 93.1 (11.1%) and 82.2% (32.4%), respectively.
Using an ivosidenib concentration-QTc model, a concentration-dependent QTc interval prolongation of approximately 17.2 msec (90% CI: 14.7, 19.7) was predicted at the steady-state Cmax based on an analysis of 173 patients with AML who received 500 mg ivosidenib once daily. A concentrationdependent QTc interval prolongation of approximately 17.2 msec (90% CI: 14.3, 20.2) was observed at the steady-state Cmax following a 500 mg daily dose based on an analysis of 101 patients with cholangiocarcinoma who received ivosidenib 500 mg daily (see sections 4.2 and 4.4).
Clinical efficacy
Newly diagnosed acute myeloid leukaemia in combination with azacitidine
The efficacy and safety of Tibsovo was evaluated in a randomised, multicenter, double-blind, placebocontrolled clinical study (AG120-C-009) of 146 adult patients with previously untreated AML with an IDH1 mutation who were ineligible for intensive induction chemotherapy, based on at least one of the following criteria: 75 years or older, Eastern Cooperative Oncology Group (ECOG) performance status of 2, severe cardiac or pulmonary disease, hepatic impairment with bilirubin >1.5 times the upper limit of normal, creatinine clearance <45 mL/min, or other comorbidity. Gene mutation analysis for central confirmation of IDH1 mutation from bone marrow and/or peripheral blood were conducted for all subjects using the Abbott RealTime IDH1 Assay. Patients were randomised to receive either Tibsovo 500 mg or matched placebo orally once daily with azacitidine 75 mg/m²/day subcutaneously or intravenously for 1 week every 4 weeks until the end of the study, disease progression or unacceptable toxicity.
The median age of patients treated with Tibsovo was 76 years (range: 58 to 84); 58% were male; 21% Asian, 17% were White, 61% not reported; and had an ECOG performance status of 0 (19%), 1 (44%), or 2 (36%). Seventy-five percent of patients had de novo AML. Overall, patients had documented favourable (4%), intermediate (67%) or poor/other (26%) cytogenetic risk as assessed by investigators based on the National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology (2017).
Efficacy was based on the primary efficacy endpoint event-free survival (EFS), measured from the date of randomisation until treatment failure, relapse from remission, or death by any cause. Treatment failure was defined as failure to achieve complete remission (CR) by week 24. Overall survival (OS), CR rate, CR + CR with partial hematologic recovery (CR + CRh) rate and objective response rate (ORR) were key secondary efficacy endpoints (Table 4 and Figure 1).
Table 4. Efficacy results in patients with newly diagnosed AML in combination with azacitidine:
| Endpoint | Ivosidenib (500 mg daily) + azacitidine N=72 | Placebo + azacitidine N=74 |
|---|---|---|
| Event-free survival, events (%) Treatment failure Relapse Death | 46 (63.9) 42 (58.3) 3 (4.2) 1 (1.4) | 62 (83.8) 59 (79.7) 2 (2.7) 1 (1.4) |
| Hazard ratio1 (95% CI) | 0.33 (0.16, 0.69) | |
| OS events (%) | 28 (38.9) | 46 (62.2) |
| Median OS (95% CI) months | 24.0 (11.3, 34.1) | 7.9 (4.1, 11.3) |
| Hazard ratio1 (95% CI) | 0.44 (0.27, 0.73) | |
| CR, n (%) | 34 (47.2) | 11 (14.9) |
| 95% CI2 | (35.3, 59.3) | (7.7, 25.0) |
| Odds ratio3 (95% CI) | 4.76 (2.15, 10.50) | |
| CR + CRh rate, n (%) | 38 (52.8) | 13 (17.6) |
| 95% CI2 | (40.7, 64.7) | (9.7, 28.2) |
| Odds ratio3 (95% CI) | 5.01 (2.32, 10.81) | |
| CR + CRi rate, n (%) | 39 (54.2) | 12 (16.2) |
| 95% CI2 | (42.0, 66.0) | (8.7, 26.6) |
| Odds ratio3 (95% CI) | 5.90 (2.69, 12.97) | |
CI: confidence interval; CR = Complete remission; CRh = Complete remission with partial hematologic recovery; CRi = Complete remission with incomplete hematologic recovery; OS = Overall survival; PR = Partial response.
1 Hazard ratio is estimated using a Cox’s proportional hazards model stratified by the randomisation stratification factors (AML status and geographic region) with PBO+AZA as the denominator.
2 CI of percentage is calculated with the Clopper and Pearson (exact Binomial) method.
3 Cochran-Mantel-Haenszel (CMH) estimate for odds ratio is calculated with PBO+AZA as the denominator.
Figure 1. Kaplan Meier plot of overall survival (OS):
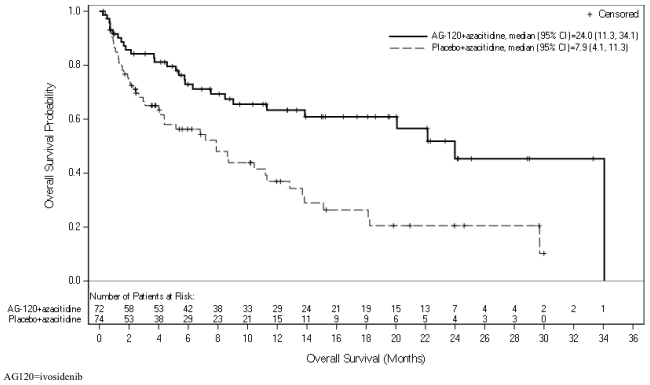
An updated OS analysis, carried out at 64.2% (N=95) of events, confirmed the overall survival benefit of Tibsovo in combination with azacitidine compared to placebo in combination with azacitidine with a median OS of 29.3 months vs 7.9 months, respectively (HR = 0.42; 95% CI: 0.27 to 0.65).
Previously treated, locally advanced or metastatic cholangiocarcinoma
The efficacy of Tibsovo was evaluated in a randomised (2:1), multicenter, double-blind, placebocontrolled, phase 3 clinical trial (Study AG120-C-005) of 185 adult patients with locally advanced or metastatic cholangiocarcinoma with an IDH1 R132 mutation whose disease had progressed following at least 1 but not more than 2 prior treatment regimens including at least one gemcitabine- or 5-FUcontaining regimen and an expected survival of ≥3 months.
Patients were randomised to receive either Tibsovo 500 mg orally once daily or matched placebo until disease progression or development of unacceptable toxicity. Randomisation was stratified by number of prior therapies (1 or 2). Eligible patients who were randomised to placebo were allowed to cross over to receive Tibsovo after documented radiographic disease progression as assessed by the Investigator. Gene mutation analysis for central confirmation of IDH1 mutation from tumour tissue biopsy were conducted on all subjects using the OncomineTM Dx Target Test.
The median age was 62 years (range: 33 to 83). Majority of patients were female (63%), 57% were White and 37% had an ECOG performance status of 0 (37%) or 1 (62%). All patients received at least 1 prior line of systemic therapy and 47% received two prior lines. Most patients had intrahepatic cholangiocarcinoma (91%) at diagnosis and 92% had metastatic disease. Across both arms, 70% patients had an R132C mutation, 15% had an R132L mutation, 12% had an R132G mutation, 1.6% had an R132S mutation, and 1.1% had an R132H mutation.
The primary efficacy outcome measure was progression free survival (PFS) as determined by Independent Radiology Center (IRC) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1, which was defined as time from randomisation to disease progression or death due to any cause.
Overall survival (OS) was a secondary efficacy endpoint. As allowed per protocol, a large proportion (70.5%) of patients in the placebo arm crossed over to receive Tibsovo following radiographic disease progression as assessed by the Investigator.
Efficacy results are summarised in Table 5.
Table 5. Efficacy results in patients with locally advanced or metastatic cholangiocarcinoma:
| Endpoint | Ivosidenib (500 mg daily) | Placebo |
|---|---|---|
| Progression-free survival (PFS) by IRC assessment | N=124 | N=61 |
| Events, n (%) Progressive Disease Death | 76 (61) 64 (52) 12 (10) | 50 (82) 44 (72) 6 (10) |
| Median PFS, months (95% CI) | 2.7 (1.6, 4.2) | 1.4 (1.4, 1.6) |
| Hazard ratio (95% CI)1 P-value2 | 0.37 (0.25, 0.54) <0.0001 | |
| PFS rate (%)3 6 months 12 months | 32.0 21.9 | NE NE |
| Ivosidenib (500 mg daily) | Placebo | |
| Overall survival4 | N=126 | N=61 |
| Deaths, n (%) | 100 (79) | 50 (82) |
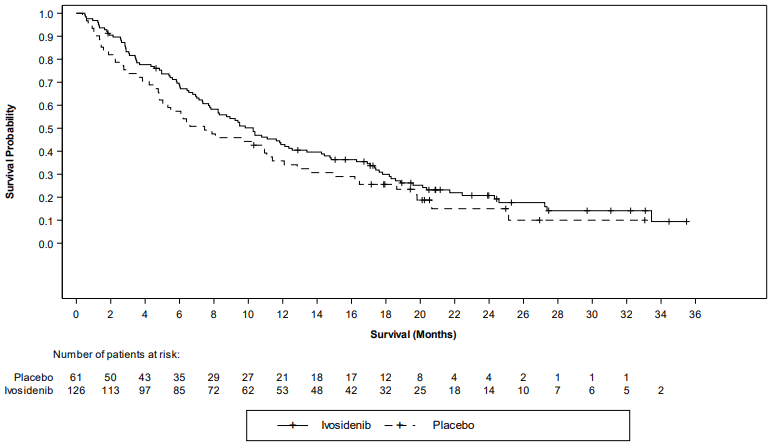
| Median OS (months, 95% CI) | 10.3 (7.8, 12.4) | 7.5 (4.8, 11.1) |
| Hazard ratio (95% CI)1 P-value2 | 0.79 (0.56, 1.12) 0.093 | |
IRC: Independent Radiology Center; CI: Confidence Interval; NE = not estimable.
1 Hazard ratio is calculated from stratified Cox regression model. Stratification factor is the number of prior line of therapies at randomisation.
2 P-value is calculated from the one-sided stratified log-rank test without adjusting for crossover. Stratification factor is the number of prior line of therapies at randomisation.
3 Based on Kaplan-Meier estimation. No patients randomised to placebo achieved PFS of 6 months or longer.
4 OS results are based on the final analysis of OS (based on 150 deaths; data cut off: 30 May 2020) which occurred 16 months after the final analysis of PFS (data cut off: 31 January 2019).
Figure 2. Kaplan Meier plot of progression-free survival (PFS) per IRC:
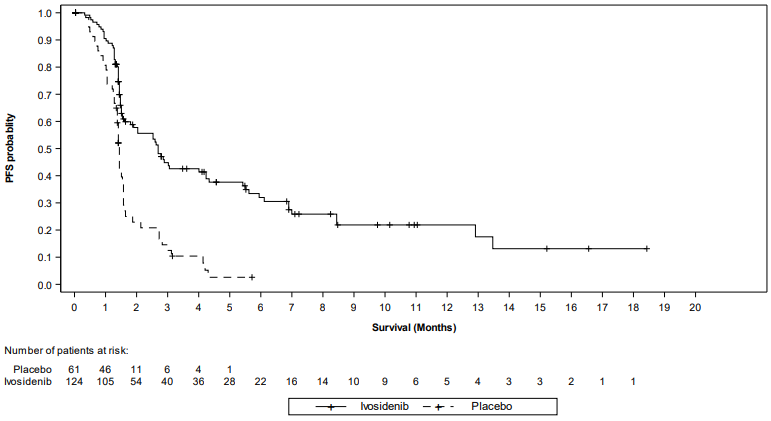
Figure 3. Kaplan-Meier plot of overall survival:
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Tibsovo in all subsets of the paediatric population in the treatment of all conditions included in the category of malignant neoplasms (except central nervous system tumours, haematopoietic and lymphoid tissue neoplasms) and in the treatment of malignant neoplasms of the central nervous system.
The European Medicines Agency has deferred the obligation to submit the results of studies with Tibsovo in one or more subsets of the paediatric population in the treatment of acute myeloid leukaemia (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
A total of 10 clinical studies have contributed to the characterisation of the clinical pharmacology of ivosidenib. Five studies have been conducted in healthy subjects and 3 studies have been conducted in patients with advanced malignancies including 2 studies in patients with cholangiocarcinoma. Two studies have been conducted in patients with newly diagnosed AML receiving ivosidenib in combination with azacitidine. Pharmacokinetic endpoints have been assessed in plasma and urine. Pharmacodynamic endpoints have been assessed in plasma, urine, tumour biopsy, and bone marrow (for studies in patients with advanced malignancies only).
The steady-state pharmacokinetics of ivosidenib 500 mg were comparable between patients with newly diagnosed AML and cholangiocarcinoma.
Absorption
After a single 500 mg oral dose, the median time to Cmax (Tmax) was approximately 2 hours in newly diagnosed AML patients treated with a combination of ivosidenib and azacitidine and in cholangiocarcinoma patients.
In patients with newly diagnosed AML treated with a combination of ivosidenib (500 mg daily dose) and azacitidine, the mean steady-state Cmax was 6,145 ng/mL (CV%: 34) and the mean steady-state AUC was 106,326 ng hr/mL (CV%: 41).
In patients with cholangiocarcinoma, the mean Cmax was 4,060 ng/mL (CV: 45) after a single dose of 500 mg and 4,799 ng/mL (CV: 33) at steady state for 500 mg daily. The AUC was 86,382 ng·hr/mL (CV%: 34).
Accumulation ratios were approximately 1.6 for AUC and 1.2 for Cmax in patients with newly diagnosed AML treated with a combination of ivosidenib and azacitidine and approximately 1.5 for AUC and 1.2 for Cmax in patients with cholangiocarcinoma, over one month, when ivosidenib was administered at 500 mg daily. Steady-state plasma levels were reached within 14 days of once daily dosing.
Significant increases in ivosidenib Cmax (by approximately 98%; 90% CI: 79, 119) and AUCinf (by approximately 25%) were observed following administration of a single dose with a high-fat meal (approximately 900 to 1,000 calories, 56% to 60% fat) in healthy subjects (see section 4.2).
Distribution
Based on a population pharmacokinetic analysis the mean apparent volume of distribution of ivosidenib at steady-state (Vc/F) is 3.20 L/kg (CV%: 47.8) in patients with newly diagnosed AML treated with a combination of ivosidenib and azacitidine and 2.97 L/kg (CV%: 25.9) in patients with cholangiocarcinoma treated with ivosidenib monotherapy.
Biotransformation
Ivosidenib was the predominant component (>92%) of total radioactivity in plasma from healthy subjects. It is primarily metabolised by oxidative pathways mediated largely by CYP3A4 with minor contributions by N-dealkylation and hydrolytic pathways.
Ivosidenib induces CYP3A4 (including its own metabolism), CYP2B6, CYP2C8, CYP2C9, and may induce CYP2C19 and UGTs. Therefore, it may decrease systemic exposure to substrates of these enzymes (see sections 4.4, 4.5 and 4.6).
Ivosidenib inhibits P-gp in vitro and has the potential to induce P-gp. Therefore, it may alter systemic exposure to active substances that are predominantly transported by P-gp (see sections 4.3 and 4.5).
In vitro data suggest that ivosidenib has the potential to inhibit OAT3, OATP1B1 and OATP1B3 at clinically relevant concentrations and it may, therefore, increase systemic exposure to OAT3, OATP1B1 or OATP1B3 substrates (see sections 4.5).
Elimination
In patients with newly diagnosed AML treated with a combination of ivosidenib and azacitidine, the mean apparent clearance of ivosidenib at steady state was 4.6 L/hour (35%) with a mean terminal halflife of 98 hours (42%).
In patients with cholangiocarcinoma, the mean apparent clearance of ivosidenib at steady state was 6.1 L/hour (31%) with a mean terminal half-life of 129 hours (102%).
In healthy subjects, 77% of a single ivosidenib oral dose was found in the faeces of which 67% was recovered unchanged. Approximately 17% of a single oral dose was found in the urine of which 10% was recovered unchanged.
Linearity/non-linearity
The AUC and Cmax of ivosidenib increased in a less than dose proportional manner from 200 mg to 1,200 mg once daily (0.4 to 2.4 times the recommended dose).
Special populations
Elderly
No clinically meaningful effects on the pharmacokinetics of ivosidenib were observed in older patients up to 84 years. The pharmacokinetics of ivosidenib in patients 85 years of age or older is unknown (see section 4.2).
Renal impairment
No clinically meaningful effects on the pharmacokinetics of ivosidenib were observed in patients with mild or moderate renal impairment (eGFR ≥30 mL/min/1.73 m²). The pharmacokinetics of ivosidenib in patients with severe renal impairment (eGFR <30 mL/min/1.73 m²) or renal impairment requiring dialysis are unknown (see section 4.2).
Hepatic impairment
Using the NCI classification, no clinically meaningful effects on the pharmacokinetics of ivosidenib were observed in patients with mild hepatic impairment. The pharmacokinetics of ivosidenib in patients with moderate and severe hepatic impairment are unknown in patients with newly diagnosed AML and with cholangiocarcinoma (see section 4.2). No PK data in patients with hepatic impairment stratified by the Child-Pugh classification are available.
Other
No clinically meaningful effects on the pharmacokinetics of ivosidenib were observed based on gender, race, body weight or ECOG performance status.
5.3. Preclinical safety data
Safety pharmacology
The potential of ivosidenib for QT prolongation was evidenced in in vitro and in vivo preclinical studies at clinically relevant plasma levels.
Repeat-dose toxicity
In animal studies at clinically relevant exposures, ivosidenib induced haematologic abnormalities (bone marrow hypocellularity, lymphoid depletion, decreased red cell mass together with extramedullary haematopoiesis in the spleen), gastrointestinal toxicity, thyroid findings (follicular cell hypertrophy/hyperplasia in rats), liver toxicity (elevated transaminases, increased weights, hepatocellular hypertrophy and necrosis in rats and hepatocellular hypertrophy associated with increased liver weights in monkeys) and kidney findings (tubular vacuolation and necrosis in rats). Toxic effects observed on haematologic system, GI system and kidney were reversible whereas the toxic effects observed on liver, spleen and thyroid were still observed at the end of the recovery period.
Genotoxicity and carcinogenicity
Ivosidenib was not mutagenic or clastogenic in conventional in vitro and in vivo genotoxicity assays. Carcinogenicity studies have not been conducted with ivosidenib.
Reproductive and developmental toxicity
Fertility studies have not been conducted with ivosidenib. In the 28-day repeat dose toxicity study in rats, uterine atrophy was observed in females at non-tolerated dose levels approximately 1.7-fold the clinical exposure (based on AUC) and was reversible after a 14-day recovery period. Testicular degeneration was observed in males at non-tolerated dose levels approximately 1.2-fold the clinical exposure (based on AUC) in animals prematurely euthanized.
In embryofoetal development studies in rats, lower foetal body weights and delayed skeletal ossification occurred in the absence of maternal toxicity. In rabbits, maternal toxicity, spontaneous abortions, decreased foetal body weights, increased post implantation loss, delayed skeletal ossification and visceral development variation (small spleen) were observed. Animal studies indicate that ivosidenib crosses the placenta and is found in foetal plasma. In rats and rabbits, the no adverse effect levels for embryofoetal development were 0.4-fold and 1.4-fold the clinical exposure (based on AUC), respectively.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.