TIVDAK Solution for injection Ref.[109850] Active ingredients: Tisotumab vedotin
Source: FDA, National Drug Code (US) Revision Year: 2024
12. Clinical Pharmacology
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of TIVDAK or of other tisotumab vedotin products.
In patients with recurrent or metastatic cervical cancer who were treated with TIVDAK 2 mg/kg every 3 weeks for up to 19 months (innovaTV 301) and 40 months (innovaTV 204), the incidence of anti-tisotumab vedotintftv antibody formation was 5.7% (12/211) and 5.4% (5/93), respectively. The incidence of neutralizing antitisotumab vedotin-tftv antibody formation was 0% (0/211) in innovaTV 301 using cell-based assay, and the incidence was 2.2% (2/93) in innovaTV 204 using Enzyme-linked Immunosorbent Assay (ELISA)-based assay. Given the low number of patients who developed anti-tisotumab vedotin-tftv antibodies, no conclusions can be drawn concerning a potential effect of immunogenicity on pharmacokinetics, efficacy or safety.
12.1. Mechanism of Action
Tisotumab vedotin-tftv is a tissue factor (TF)-directed antibody drug conjugate (ADC). The antibody is a human IgG1 directed against cell surface TF. TF is the primary initiator of the extrinsic blood coagulation cascade. The small molecule, MMAE, is a microtubule-disrupting agent, attached to the antibody via a protease-cleavable linker. Nonclinical data suggests that the anticancer activity of tisotumab vedotin-tftv is due to the binding of the ADC to TF expressing cancer cells, followed by internalization of the ADC-TF complex, and release of MMAE via proteolytic cleavage. MMAE disrupts the microtubule network of actively dividing cells, leading to cell cycle arrest and apoptotic cell death. In vitro, tisotumab vedotin-tftv also mediates antibody-dependent cellular phagocytosis and antibody-dependent cellular cytotoxicity.
12.2. Pharmacodynamics
Tisotumab vedotin-tftv exposure-response relationships and the time course of pharmacodynamics response have not been fully characterized.
Cardiac Electrophysiology
At the recommended dose, tisotumab vedotin-tftv had no large mean effect on QTc prolongation (>20 msec).
12.3. Pharmacokinetics
Table 8 summarizes the exposure parameters of tisotumab vedotin-tftv and unconjugated MMAE (the cytotoxic component of tisotumab vedotin-tftv) following administration of one 3-week cycle of tisotumab vedotin-tftv 2 mg/kg to patients. Tisotumab vedotin-tftv concentrations peaked near the end of the infusion, while unconjugated MMAE concentrations peaked approximately 2 to 3 days after tisotumab vedotin-tftv dosing. Tisotumab vedotin-tftv Cmax increased proportionally, while AUC0-last increased in a more than dose-proportional manner, after a single dose ranging from 0.3–2.2 mg/kg (0.15 to 1.1 times the approved recommended dose). There was no accumulation of tisotumab vedotin-tftv and unconjugated MMAE. Steadystate concentrations of tisotumab vedotin-tftv and unconjugated MMAE were reached after 1 treatment cycle.
Table 8. Exposure Parameters of Tisotumab Vedotin-tftv and Unconjugated MMAE:
| Tisotumab Vedotin-tftv Mean (± SD) | Unconjugated MMAE Mean (± SD) | |
|---|---|---|
| Cmax | 40.8 (8.12) μg/mL | 5.91 (4.2) ng/mL |
| AUC | 57.5 (13.4) day*μg/mL | 50 (35.8) day*ng/mL |
Cmax = maximum concentration, AUC = area under the concentration-time curve from time 0 to 21 days (3 weeks)
Distribution
The tisotumab vedotin-tftv steady state volume of distribution is 7.83 (%CV: 19.1) L. Plasma protein binding of MMAE ranged from 68% to 82%, in vitro.
Elimination
The median terminal half-life of tisotumab vedotin-tftv and unconjugated MMAE is 4.04 (range: 2.26-7.25) days and 2.56 (range: 1.81-4.10) days, respectively. The linear clearance of tisotumab vedotin-tftv and unconjugated MMAE was 1.54 (%CV: 28.8) L/day and 45.9 (%CV: 61.1) L/day, respectively. Elimination of MMAE appeared to be limited by its rate of release from tisotumab vedotin-tftv.
Metabolism
Tisotumab vedotin-tftv is expected to undergo catabolism to small peptides, amino acids, unconjugated MMAE, and unconjugated MMAE-related catabolites. Tisotumab vedotin-tftv releases unconjugated MMAE via proteolytic cleavage, and unconjugated MMAE is primarily metabolized by CYP3A4 in vitro.
Excretion
The excretion of tisotumab vedotin-tftv is not fully characterized. Following a single-dose of another ADC that contains MMAE, 17% of the total MMAE administered was recovered in feces and 6% in urine over a 1-week period, primarily as unchanged drug. A similar excretion profile of MMAE is expected after tisotumab vedotintftv administration.
Specific Populations
No clinically significant differences in the pharmacokinetics of tisotumab vedotin-tftv were observed based on age (21 to 81 years), sex, race (white vs non-white) or ethnicity (Hispanic or Latino vs non-Hispanic or nonLatino). No clinically significant differences in exposures of tisotumab vedotin-tftv and unconjugated MMAE were observed in patients with mild to moderate renal impairment (CLcr 30 to <90 mL/min using the Cockcroft-Gault equation) compared to patients with normal renal function. The effect of severe renal impairment (CLcr 15 to <30 mL/min) or end-stage renal disease with or without dialysis on pharmacokinetics of tisotumab vedotin-tftv and unconjugated MMAE is unknown.
Patients with Hepatic Impairment
Unconjugated MMAE exposures were 37% higher, but there were no clinically significant differences in exposures of tisotumab vedotin-tftv in patients with mild hepatic impairment compared to patients with normal hepatic function. The effect of moderate or severe hepatic impairment or liver transplantation on the pharmacokinetics of tisotumab vedotin-tftv or unconjugated MMAE is unknown.
Drug Interaction Studies
Clinical Studies
No clinical studies evaluating the drug-drug interaction potential of tisotumab vedotin-tftv have been conducted. To characterize the drug-drug interaction potential of unconjugated MMAE, clinical studies with another ADC that contains MMAE are described below, and similar effects on tisotumab vedotin-tftv and unconjugated MMAE exposures are expected with concomitant use of TIVDAK.
There were no clinically significant differences in midazolam (sensitive CYP3A4 substrate) pharmacokinetics when used concomitantly with another ADC that contains MMAE.
Strong CYP3A4 Inhibitors: Ketoconazole (strong CYP3A4 inhibitor) used concomitantly with another ADC that contains MMAE increased unconjugated MMAE Cmax by 25% and AUC by 34%, with no change in ADC exposure.
Strong CYP3A4 Inducers: Rifampin (strong CYP3A4 inducer) used concomitantly with another ADC that contains MMAE decreased unconjugated MMAE Cmax by 44% and AUC by 46%, with no change in ADC exposure.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: MMAE does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6. MMAE did not induce any major CYP450 enzymes in human hepatocytes.
Transporter Systems: MMAE is a substrate of P-glycoprotein (P-gp), but not an inhibitor of P-gp.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies in animals have not been performed with tisotumab vedotin-tftv or MMAE.
MMAE was positive for genotoxicity in the in vivo rat bone marrow micronucleus study through an aneugenic mechanism. MMAE was not mutagenic in the bacterial reverse mutation (Ames) assay or the L5178 TK+/- mouse lymphoma forward mutation assay.
Fertility studies with tisotumab vedotin-tftv or MMAE have not been conducted. However, results of a repeatdose toxicity study in monkeys indicate the potential for tisotumab vedotin-tftv to impair male reproductive function and fertility.
In a repeat-dose toxicology study conducted in monkeys for 13 weeks, doses ≥1 mg/kg tisotumab vedotin-tftv (≥0.6 times the human exposure [AUC] at the recommended dose) resulted in decreased testicular size and seminiferous tubule atrophy, reduction or absence in sperm count, and decreased sperm motility. Findings of sperm absence and decreased motility did not reverse by the end of the recovery period at doses ≥3 mg/kg (≥1.7 times the human exposure [AUC] at the recommended dose).
MMAE-containing ADCs have been associated with adverse ovarian effects when administered to sexually immature animals. Adverse effects included decrease in, or absence of, secondary and tertiary ovarian follicles after weekly administration to cynomolgus monkeys in studies of 4-week duration. These effects showed a trend towards recovery 6 weeks after the end of dosing; no changes were observed in primordial follicles.
14. Clinical Studies
14.1 Recurrent or Metastatic Cervical Cancer
innovaTV 301
The efficacy of TIVDAK was evaluated in innovaTV 301 (NCT04697628), an open-label, active-controlled, multicenter, randomized trial that enrolled 502 patients with recurrent or metastatic cervical cancer who had received one or two prior systemic therapy regimens in the recurrent or metastatic setting, including chemotherapy with or without bevacizumab and/or an anti-PD-(L)1 agent. Patients were excluded if they had active ocular surface disease, any prior episode of cicatricial conjunctivitis or ocular SJS, Grade ≥2 peripheral neuropathy, or clinically significant bleeding issues or risks.
Patients were randomized (1:1) to receive either TIVDAK 2 mg/kg intravenously every 3 weeks (n=253) or investigator's choice of chemotherapy (n=249) consisting of topotecan, vinorelbine, gemcitabine, irinotecan or pemetrexed, until unacceptable toxicity or disease progression. Randomization was stratified by prior treatment with bevacizumab (yes or no), prior anti-PD-(L)1 therapy (yes or no), region (US, Europe, or Other), and ECOG performance status (0 or 1).
Patients were treated until disease progression or unacceptable toxicity. Tumor response assessments were performed every 6 weeks for the first 30 weeks and every 12 weeks thereafter.
The major efficacy outcome measure was overall survival (OS). Additional efficacy outcome measures were progression free survival (PFS) and confirmed objective response rate (ORR) as assessed by investigator using RECIST v1.1.
The median age was 50 years (range: 26 to 80); 49% were White, 36% were Asian, 10% were not reported, 3% were American Indian or Alaskan Native, and 2% were Black; 20% were Hispanic/Latino; and baseline ECOG performance status was 0 (54%) or 1 (46%). Sixty-three percent of patients had squamous cell carcinoma, 32% had adenocarcinoma, and 5% had adenosquamous histology. Ninety percent of patients had extrapelvic disease; 61% of patients had received 1 prior line of systemic therapy, and 38% had 2 prior lines of systemic therapy. All patients received prior chemotherapy; 64% received prior bevacizumab and 27% received prior anti-PD-1 or anti-PD-L1 therapy. Patients on the control arm received gemcitabine (44%), pemetrexed (32%), topotecan (8%), vinorelbine (7%), or irinotecan (6%).
Statistically significant improvements in OS, PFS, and ORR were demonstrated for TIVDAK compared with chemotherapy.
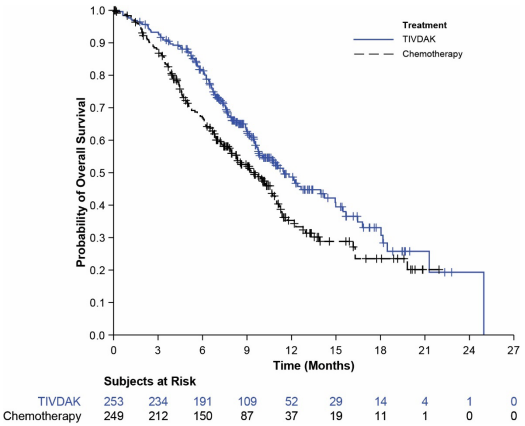
Table 9 and Figure 2 summarize the efficacy results from innovaTV 301.
Table 9. Efficacy Results in innovaTV 301:
| Endpoint | TIVDAK N=253 | Chemotherapy N=249 |
|---|---|---|
| 3< Overall Survival | ||
| Number (%) of patients with events | 123 (48.6) | 140 (56.2) |
| Median in months (95% CI) | 11.5 (9.8, 14.9) | 9.5 (7.9, 10.7) |
| Hazard ratio (95% CI) | 0.70 (0.54, 0.89) | |
| p-value | 0.00381 | |
| Progression Free Survival | ||
| Number (%) of patients with events | 198 (78.3) | 194 (77.9) |
| Median in months (95% CI) | 4.2 (4.0, 4.4) | 2.9 (2.6, 3.1) |
| Hazard ratio (95% CI) | 0.67 (0.54, 0.82) | |
| p-value | <0.00012 | |
| Confirmed Objective Response Rate (CR + PR) | ||
| ORR (%) (95% CI) | 17.8 (13.3, 23.1) | 5.2 (2.8, 8.8) |
| p-value | <0.00013 | |
| Complete response rate (%) | 2.4 | 0 |
| Partial response rate (%) | 15.4 | 5.2 |
CI: confidence interval
1 Based on a stratified log-rank test. The threshold for statistical significance is 0.0226 (2-sided).
2 Based on a stratified log-rank test. The threshold for statistical significance is 0.0453 (2-sided).
3 Based on CMH test. The threshold for statistical significance is 0.05 (2-sided).
Figure 2. Kaplan Meier Plot of Overall Survival:
innovaTV 204
The efficacy of TIVDAK was evaluated in innovaTV 204 (NCT03438396), an open-label, multicenter, singlearm trial that treated 101 patients with recurrent or metastatic cervical cancer who had received no more than two prior systemic regimens in the recurrent or metastatic setting, including at least one prior platinum-based chemotherapy regimen. Patients were excluded if they had active ocular surface disease, any prior episode of cicatricial conjunctivitis or ocular SJS, Grade ≥2 peripheral neuropathy or known coagulation defects leading to an increased risk of bleeding.
Patients received TIVDAK 2 mg/kg intravenously every 3 weeks until disease progression or unacceptable toxicity. Tumor response assessments were performed every 6 weeks for the first 30 weeks and every 12 weeks thereafter.
The median age was 50 years (range: 31 to 78); 95% were White, 2% were Asian, and 1% were Black. Six percent of patients were Hispanic or Latino. Sixty-eight percent of patients had squamous cell carcinoma, 27% had adenocarcinoma, and 5% had adenosquamous histology. ECOG performance status was 0 (58%) or 1 (42%). Seventy percent of patients had received 1 prior line of systemic therapy, and 30% had 2 prior lines of systemic therapy. Sixty-nine percent of patients previously received bevacizumab as part of their prior systemic therapy. Sixty-three percent received bevacizumab in combination with chemotherapy (paclitaxel and cisplatin or carboplatin, or paclitaxel and topotecan) as first-line therapy.
The major efficacy outcome measures were confirmed objective response rate (ORR) as assessed by an independent review committee (IRC) using RECIST v1.1 criteria and duration of response (DOR).
Efficacy results are presented in Table 10.
Table 10. Efficacy Results in innovaTV 204 by IRC:
| Endpoint | N=101 |
|---|---|
| Confirmed ORR (95% CI) | 24% (15.9, 33.3) |
| Complete response rate | 7% |
| Partial response rate | 17% |
| Duration of Response | |
| Median Duration of Response, months1 (95% CI) | 8.3 (4.2, NR) |
1 Based on patients (n=24) with a response by IRC.
CI: confidence interval
NR: not reached
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.