TRIVERAM Film-coated tablet Ref.[107808] Active ingredients: Amlodipine Atorvastatin Atorvastatin, Amlodipine and Perindopril Perindopril
Source: Health Sciences Authority (SG) Revision Year: 2023 Publisher: <u>France:</u> Les Laboratoires Servier, 50, rue Carnot, 92284 Suresnes cedex, France <u>Singapore:</u> Servier (S) Pte Ltd, 67 Ubi Avenue 1, #06-09 StarHub Green, Singapore 408942
4.3. Contraindications
- Hypersensitivity to the active substances or to any other ACE inhibitor or dihydropyridine derivatives or statin or to any of the excipients of this medicinal product listed in section 6.1;
- Active liver disease or unexplained persistent elevations of serum transaminases exceeding 3 times the upper limit of normal;
- During pregnancy, while breast-feeding and in women of child-bearing potential not using appropriate contraceptive measures (see section 4.6);
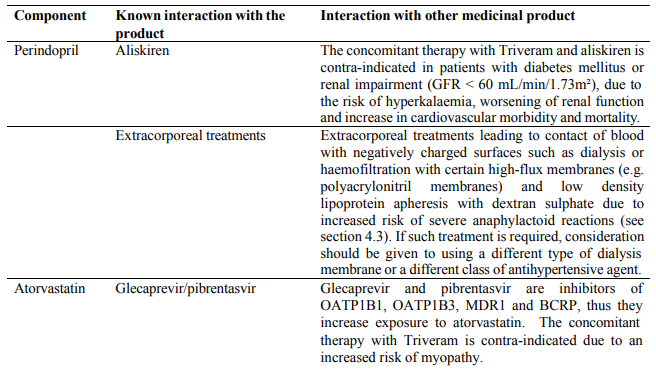
- Concomitant use with the hepatitis C antivirals glecaprevir/pibrentasvir;
- Severe hypotension;
- Shock (including cardiogenic shock);
- Obstruction of the outflow tract of the left ventricle (e.g., hypertrophic obstructive cardiomyopathy and high grade aortic stenosis);
- Haemodynamically unstable heart failure after acute myocardial infarction;
- History of angioedema (Quincke’s oedema) associated with previous ACE inhibitor therapy;
- Hereditary or idiopathic angioedema;
- Concomitant use with aliskiren-containing products in patients with diabetes mellitus or renal impairment (GFR <60 mL/min/1.73m²) (see sections 4.5 and 5.1);
- Concomitant use with sacubitril/valsartan therapy. Triveram must not be initiated earlier than 36 hours after the last dose of sacubitril/valsartan (see sections 4.4 and 4.5);
- Extracorporeal treatments leading to contact of blood with negatively charged surfaces (see section 4.5); Significant bilateral renal artery stenosis or stenosis of the artery to a single functioning kidney (see section 4.4).
4.4. Special warnings and precautions for use
Special warnings and precautions related to atorvastatin, perindopril and amlodipine are applicable to Triveram
Hepatic impairment
Due to the atorvastatin component in Triveram, liver function tests should be performed periodically. Patients who develop any signs or symptoms suggestive of hepatic dysfunction should have liver function tests performed. Patients who develop increased transaminase levels should be monitored until the abnormality(ies) resolve. Should an increase in transaminases of greater than 3 times the upper limit of normal (ULN) persist, reduction of atorvastatin dose using the individual components or withdrawal of atorvastatin is recommended (see section 4.8). Triveram should be used with caution in patients who consume substantial quantities of alcohol and/or have a history of liver disease.
Rarely, ACE inhibitors have been associated with a syndrome that starts with cholestatic jaundice and progresses to fulminant hepatic necrosis and (sometimes) death. The mechanism of this syndrome is not understood. Patientsreceiving Triveram who develop jaundice or marked elevations of hepatic enzymesshould discontinue Triveram and receive appropriate medical follow-up (see section 4.8).
The half life of amlodipine is prolonged and AUC values are higher in patients with impaired liver function; dosage recommendations have not been established. Careful monitoring may be required in patients treated with Triveram and suffering from severe hepatic impairment.
Taking into account the effect of atorvastatin, perindopril and amlodipine, Triveram is contra-indicated in patients with active liver disease or unexplained persistent elevations of serum transaminases exceeding 3 times the upper limit of normal. Triveram should be used with caution in patients with hepatic impairment and in patients who consume substantial quantities of alcohol and/or have a history of liver disease. If a change of posology is required, titration should be done with the individual components.
Skeletal muscle effects
Atorvastatin, like other HMG-CoA reductase inhibitors, may in rare occasions affect the skeletal muscle and cause myalgia, myositis, and myopathy that may progress to rhabdomyolysis, a potentially life-threatening condition characterised by markedly elevated creatine kinase (CK) levels (>10 times ULN), myoglobinaemia and myoglobinuria which may lead to renal failure.
There have been very rare reports of an immune-mediated necrotising myopathy (IMNM) during or after treatment with some statins. IMNM is clinically characterised by persistent proximal muscle weakness and elevated serum creatine kinase, which persist despite discontinuation of statin treatment, positive anti-HMG CoA reductase antibody and improvement with immunosuppressive agents.
Creatine kinase measurement
Creatine kinase (CK) should not be measured following strenuous exercise or in the presence of any plausible alternative cause of CK increase as this makes value interpretation difficult. If CK levels are significantly elevated at baseline (> 5 times ULN), levels should be remeasured within 5 to 7 days later to confirm the results.
Before the treatment
Atorvastatin should be prescribed with caution in patients with pre-disposing factors for rhabdomyolysis. A CK level should be measured before starting statin treatment in the following situations:
- Renal impairment
- Hypothyroidism
- Personal or familial history of hereditary muscular disorders
- Previous history of muscular toxicity with a statin or fibrate
- Previous history of liver disease and/or where substantial quantities of alcohol are consumed
- In elderly (age >70 years), the necessity of such measurement should be considered, according to the presence of other predisposing factors for rhabdomyolysis
- Situations where an increase in plasma levels may occur, such as interactions (see section 4.5) and special populations including genetic subpopulations (see section 5.2)
In such situations, the risk of treatment should be considered in relation to possible benefit, and clinical monitoring is recommended. If CK levels are significantly elevated (>5 times ULN) at baseline, treatment should not be started.
Whilst on treatment:
- Patients must be asked to promptly report muscle pain, cramps, or weakness especially if accompanied by malaise or fever.
- If such symptoms occur whilst a patient is receiving treatment with Triveram, their CK levels should be measured. If these levels are found to be significantly elevated (>5 times ULN), treatment should be stopped.
- If muscular symptoms are severe and cause daily discomfort, even if the CK levels are elevated to ≤5 x ULN, treatment discontinuation should be considered.
- If symptoms resolve and CK levels return to normal, then re-introduction of atorvastatin or introduction of an alternative statin may be considered at the lowest dose and with close monitoring.
- Triveram must be discontinued immediately if clinically significant elevation of CK levels (>10 x ULN) occur, or if rhabdomyolysis is diagnosed or suspected.
Concomitant treatment with other medicinal products
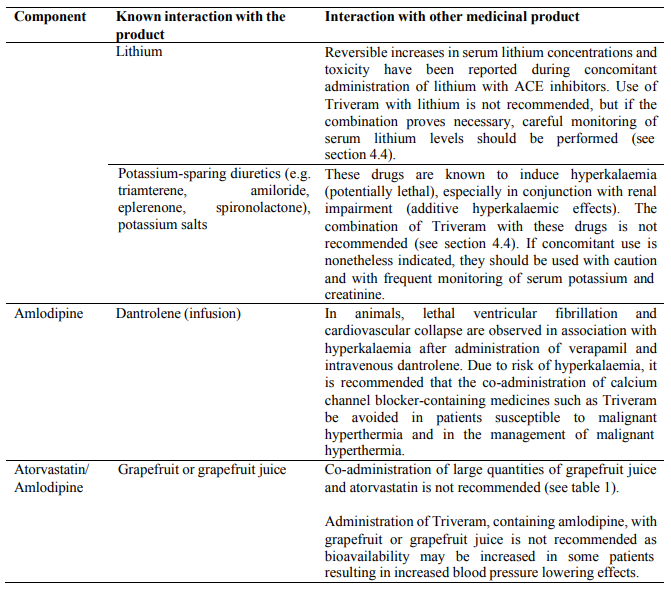
Due to atorvastatin component, risk of rhabdomyolysis is increased when Triveram is administered concomitantly with certain medicinal products that may increase the plasma concentration of atorvastatin such as potent inhibitors of CYP3A4 or transport proteins (e.g. ciclosporine, telithromycin, clarithromycin, delavirdine, stiripentol, ketoconazole, voriconazole, itraconazole, posaconazole, letermovir and HIV protease inhibitors including ritonavir, lopinavir, atazanavir, indinavir, darunavir, tipranavir/ritonavir, etc). The risk of myopathy may also be increased with the concomitant use of gemfibrozil and other fibric acid derivatives, antivirals for the treatment of hepatitis C (HCV) (boceprevir, telaprevir, elbasvir/grazoprevir, ledipasvir/sofosbuvir), erythromycin, niacin or ezetimibe. If possible, alternative (non-interacting) therapies should be considered instead of these medicinal products.
In cases where co-administration of these medicinal products with Triveram is necessary, the benefit and the risk of concurrent treatment should be carefully considered. When patients are receiving medicinal products that increase the plasma concentration of atorvastatin, a lower maximum dose of atorvastatin is recommended, hence down-titration with the individual components should be considered. In addition, in the case of potent CYP3A4 inhibitors, a lower starting dose of atorvastatin should be considered and appropriate clinical monitoring of these patients is recommended (see section 4.5).
Due to atorvastatin component, Triveram must not be co-administered with systemic formulations of fusidic acid or within 7 days of stopping fusidic acid treatment. In patients where the use of systemic fusidic acid is considered essential, statin treatment should be discontinued throughout the duration of fusidic acid treatment. There have been reports of rhabdomyolysis (including some fatalities) in patients receiving fusidic acid and statins in combination (see section 4.5). The patient should be advised to seek medical advice immediately if they experience any symptoms of muscle weakness, pain or tenderness.
Statin therapy may be re-introduced seven days after the last dose of fusidic acid.
In exceptional circumstances, where prolonged systemic fusidic acid is needed, e.g., for the treatment of severe infections, the need for co-administration of Triveram and fusidic acid should only be considered on a case by case basis and under close medical supervision.
Myasthenia gravis, ocular myasthenia
In few cases, statins have been reported to induce de novo or aggravate pre-existing myasthenia gravis or ocular myasthenia (see section 4.8). Triveram should be discontinued in case of aggravation of symptoms. Recurrences when the same or a different statin was (re-) administered have been reported.
Interstitial lung disease
Exceptional cases of interstitial lung disease have been reported with some statins, especially with long term therapy (see section 4.8). Presenting features can include dyspnoea, non-productive cough and deterioration in general health (fatigue, weight loss and fever). If it is suspected a patient has developed interstitial lung disease, Triveram therapy should be discontinued.
Diabetes Mellitus
Some evidence suggests that statins as a class raise blood glucose and in some patients, at high risk of future diabetes, may produce a level of hyperglycaemia where formal diabetes care is appropriate. This risk, however, is outweighed by the reduction in vascular risk with statins and therefore should not be a reason for stopping Triveram treatment. Patients at risk (fasting glucose 5.6 to 6.9 mmol/L, BMI > 30kg/m², raised triglycerides, hypertension) should be monitored both clinically and biochemically according to national guidelines when treated with Triveram.
In diabetic patients treated with oral antidiabetic agents or insulin, glycaemic control should be closely monitored during the first month of treatment with medicines containing an ACE inhibitor, such as Triveram (see section 4.5).
Cardiac failure
Triveram should be used with caution in patients with heart failure. In a long-term, placebo controlled study in patients with severe heart failure (NYHA class III and IV) the reported incidence of pulmonary oedema was higher in the amlodipine treated group than in the placebo group (see section 5.1). Medicines containing calcium channel blockers, including amlodipine, should be used with caution in patients with congestive heart failure, as they may increase the risk of future cardiovascular events and mortality.
Hypotension
ACE inhibitors, such as perindopril, may cause a fall in blood pressure. Symptomatic hypotension is seen rarely in uncomplicated hypertensive patients and is more likely to occur in patients who have been volumedepleted e.g. by diuretic therapy, dietary salt restriction, dialysis, diarrhoea or vomiting, or who have severe renin-dependent hypertension (see sections 4.5 and 4.8). In patients with symptomatic heart failure, with or without associated renal insufficiency, symptomatic hypotension has been observed. This is most likely to occur in those patients with more severe degrees of heart failure, as reflected by the use of high doses of loop diuretics, hyponatraemia or functional renal impairment. In patients at increased risk of symptomatic hypotension, initiation of therapy and dose adjustment should be closely monitored (see sections 4.8). Similar considerations apply to patients who suffer from ischaemic heart or cerebrovascular disease in whom an excessive fall in blood pressure could result in a myocardial infarction or cerebrovascular accident.
If hypotension occurs, the patient should be placed in the supine position and, if necessary, should receive an intravenous infusion of sodium chloride 9 mg/mL (0.9%) solution. A transient hypotensive response is not a contraindication to further doses, which can be given usually without difficulty once the blood pressure has increased after volume expansion.
In some patients with congestive heart failure who have normal or low blood pressure, additional lowering of systemic blood pressure may occur with perindopril. This effect is anticipated and is usually not a reason to discontinue treatment. If hypotension becomes symptomatic, a reduction of dose or discontinuation of treatment with Triveram may be necessary.
Aortic and mitral valve stenosis
As with other medicines containing ACE inhibitors such as perindopril, Triveram should be given with caution to patients with mitral valve stenosis or significant aortic stenosis that is not high grade. The use of Triveram is contraindicated in patients with severe obstruction of the outflow tract of the left ventricle (see section 4.3).
Kidney transplantation
There is no experience regarding the administration of perindopril arginine in patients with a recent kidney transplantation.
Renovascular hypertension
There is an increased risk of hypotension and renal insufficiency when patient with bilateral renal artery stenosis or stenosis of the artery to a single functioning kidney are treated with ACE inhibitors (see section 4.3). Treatment with diuretics may be a contributory factor. Loss of renal function may occur with only minor changes in serum creatinine even in patients with unilateral renal artery stenosis.
Renal impairment
Triveram can be administered in patients with creatinine clearance ≥60mL/min, and is not suitable for patients with creatinine clearance <60mL/min (moderate to severe renal impairment). In these patients, an individual dose titration with the monocomponents is recommended. Routine monitoring of potassium and creatinine are part of normal medical practice for patients with renal impairment (see section 4.8).
In patients with symptomatic heart failure, hypotension following the initiation of therapy with ACE inhibitors, such as perindopril, may lead to some further impairment in renal function. Acute renal failure, usually reversible, has been reported in this situation.
In some patients with bilateral renal artery stenosis or stenosis of the artery to a solitary kidney, who have been treated with ACE inhibitors, increases in blood urea and serum creatinine, usually reversible upon discontinuation of therapy, have been seen. This is especially likely in patients with renal insufficiency. If renovascular hypertension is also present there is an increased risk of severe hypotension and renal insufficiency.
Some hypertensive patients with no apparent pre-existing renal vascular disease have developed increases in blood urea and serum creatinine, usually minor and transient, especially when perindopril has been given concomitantly with a diuretic. This is more likely to occur in patients with pre-existing renal impairment. Dosage reduction and/or discontinuation of the diuretic and/or Triveram may be required.
Amlodipine may be used at normal doses in patients with renal failure. Changes in amlodipine plasma concentration are not correlated with degree of renal impairment.
The effect of the combination Triveram has not been tested in patients with renal impairment. Triveram doses should respect the dosing recommendations of the individual components taken separately.
Haemodialysis patients
Anaphylactoid reactions have been reported in patients dialysed with high flux membranes, and treated concomitantly with an ACE inhibitor. In these patients consideration should be given to using a different type of dialysis membrane or different class of antihypertensive agent.
Hypersensitivity/Angioedema
Angioedema of the face, extremities, lips, mucous membranes, tongue, glottis and/or larynx has been reported rarely in patients treated with ACE inhibitors, including perindopril (see section 4.8). This may occur at any time during therapy. In such cases, Triveram should promptly be discontinued and appropriate monitoring should be initiated and continued until complete resolution ofsymptoms has occurred. In those instances where swelling was confined to the face and lips the condition generally resolved without treatment, although antihistamines have been useful in relieving symptoms.
Angioedema associated with laryngeal oedema may be fatal. Where there is involvement of the tongue, glottis or larynx, likely to cause airway obstruction, emergency therapy should be administered promptly. This may include the administration of adrenaline and/or the maintenance of a patent airway. The patient should be under close medical supervision until complete and sustained resolution of symptoms has occurred. Patients with a history of angioedema unrelated to ACE inhibitor therapy may be at increased risk of angioedema while receiving Triveram (see section 4.3).
Intestinal angioedema has been reported rarely in patients treated with ACE inhibitors. These patients presented with abdominal pain (with or without nausea or vomiting); in some cases there was no prior facial angioedema and C-1 esterase levels were normal. The angioedema was diagnosed by procedures including abdominal CT scan, or ultrasound or at surgery and symptoms resolved after stopping the ACE inhibitor. Intestinal angioedema should be included in the differential diagnosis of patients treated with Triveram presenting with abdominal pain.
The combination of perindopril with sacubitril/valsartan is contraindicated due to the increased risk of angioedema (see section 4.3). Sacubitril/valsartan must not be initiated until 36 hours after taking the last dose of perindopril therapy. If treatment with sacubitril/valsartan is stopped, perindopril therapy must not be initiated until 36 hours after the last dose of sacubitril/valsartan (see sections 4.3 and 4.5). Concomitant use of ACE inhibitors with NEP inhibitors (e.g. racecadotril), mTOR inhibitors (e.g. sirolimus, everolimus, temsirolimus) and gliptins (e.g. linagliptin, saxagliptin, sitagliptin, vildagliptin) may lead to an increased risk of angioedema (e.g. swelling of the airways or tongue, with or without respiratory impairment) (see section 4.5). Caution should be used when starting racecadotril, mTOR inhibitors (e.g. sirolimus, everolimus, temsirolimus) and gliptins (e.g. linagliptin, saxagliptin, sitagliptin, vildagliptin) in a patient already taking an ACE inhibitor.
Anaphylactoid reactions during low-density lipoproteins (LDL) apheresis
Rarely, patients receiving ACE inhibitors during low-density lipoprotein (LDL) apheresis with dextran sulphate have experienced life-threatening anaphylactoid reactions. These reactions were avoided by temporarily withholding ACE inhibitor therapy prior to each apheresis.
Anaphylactoid reactions during desensitisation
Patients receiving ACE inhibitor-containing medicines, such as Triveram, during desensitisation treatment (e.g. hymenoptera venom) have experienced anaphylactoid reactions. In the same patients, these reactions have been avoided when the ACE inhibitors were temporarily withheld, but they reappeared upon inadvertent rechallenge.
Neutropenia/Agranulocytosis/Thrombocytopenia/Anaemia
Neutropenia/agranulocytosis, thrombocytopenia and anaemia have been reported in patients receiving ACE inhibitors. In patients with normal renal function and no other complicating factors, neutropenia occurs rarely. Triveram should be used with extreme caution in patients with collagen vascular disease, immunosuppressant therapy, treatment with allopurinol or procainamide, or a combination of these complicating factors, especially if there is pre-existing impaired renal function. Some of these patients developed serious infections, which in a few instances did not respond to intensive antibiotic therapy. If Triveram is used in such patients, periodic monitoring of white blood cell countsis advised and patients should be instructed to report any sign of infection (e.g. sore throat, fever).
Race
ACE inhibitors cause a higher rate of angioedema in black patients than in non-black patients. Triveram, which contains the ACE inhibitor perindopril, may be less effective in lowering blood pressure in black people than in non-blacks, possibly because of a higher prevalence of low-renin states in the black hypertensive population.
Cough
Cough has been reported with the use of ACE inhibitors. Characteristically, the cough is non-productive, persistent and resolves after discontinuation of therapy. ACE inhibitor-induced cough should be considered as part of the differential diagnosis of cough in patients treated with Triveram.
Surgery/Anaesthesia
In patients undergoing major surgery or during anaesthesia with agents that produce hypotension, Triveram may block angiotensin II formation secondary to compensatory renin release. The treatment should be discontinued one day prior to the surgery. If hypotension occurs and is considered to be due to this mechanism, it can be corrected by volume expansion.
Hyperkalaemia
Elevations in serum potassium have been observed in some patients treated with ACE inhibitors, including perindopril, ACE inhibitors can cause hyperkalaemia because they inhibit the release of aldosterone. The effect is usually not significant in patients with normal renal function. Risk factors for the development of hyperkalaemia include those with renal insufficiency, worsening of renal function, age (>70 years), diabetes mellitus, intercurrent events, in particular dehydration, acute cardiac decompensation, metabolic acidosis and concomitant use of potassium-sparing diuretics (e.g. spironolactone, eplerenone, triamterene, or amiloride), potassium supplements or potassium-containing salt substitutes; or those patients taking other drugs associated with increases in serum potassium (e.g. heparin, co-trimoxazole also known as trimethoprim/sulfamethoxazole) and especially aldosterone antagonists or angiotensin-receptor blockers. The use of potassium supplements, potassium-sparing diuretics, or potassium-containing salt substitutes particularly in patients with impaired renal function may lead to a significant increase in serum potassium. Hyperkalaemia can cause serious, sometimes fatal arrhythmias. Potassium-sparing diuretics and angiotensinreceptor blockers should be used with caution in patients receiving ACE inhibitors, and serum potassium and renal function should be monitored. If concomitant use of the above-mentioned agents with Triveram is deemed appropriate, they should be used with caution and with frequent monitoring of serum potassium (see section 4.5).
Combination with lithium
The combination of lithium and medicines containing perindopril, such as Triveram, is not recommended (see section 4.5).
Dual blockade of the renin-angiotensin-aldosterone system (RAAS)
There is evidence that the concomitant use of ACE-inhibitors, angiotensin II receptor blockers or aliskiren increases the risk of hypotension, hyperkalaemia and decreased renal function (including acute renal failure). Dual blockade of RAAS through the combined use of ACE-inhibitors, angiotensin II receptor blockers or aliskiren is therefore not recommended (see sections 4.5 and 5.1).
If dual blockade therapy is considered absolutely necessary, this should only occur under specialist supervision and subject to frequent close monitoring of renal function, electrolytes and blood pressure.
ACE-inhibitors and angiotensin II receptor blockers should not be used concomitantly in patients with diabetic nephropathy.
Primary aldosteronism
Patients with primary hyperaldosteronism generally will not respond to anti-hypertensive drugs acting through inhibition of the renin-angiotensin system. Therefore, the use of this product is not recommended.
Excipients
Due to the presence of lactose, patients with rare hereditary problems of galactose intolerance, glucosegalactose malabsorption, or total lactase deficiency should not take Triveram.
Level of sodium
Triveram contains less than 1 mmol sodium (23 mg) per tablet, i.e. essentially ‘sodium-free’.
4.5. Interaction with other medicinal products and other forms of interaction
Clinical trial data has shown that dual blockade of the renin-angiotensin-aldosterone-system (RAAS) through the combined use of ACE-inhibitors, angiotensin II receptor blockers or aliskiren is associated with a higher frequency of adverse events such as hypotension, hyperkalaemia and decreased renal function (including acute renal failure) compared to the use of a single RAAS-acting agent (see sections 4.3, 4.4 and 5.1).
No drug interaction studies have been conducted with Triveram and other drugs, although studies have been conducted with atorvastatin, perindopril and amlodipine separately. The results of these studies are provided below.
Drugs increasing the risk of angioedema
Concomitant use of ACE inhibitors with sacubitril/valsartan is contraindicated as this increases the risk of angioedema (see section 4.3 and 4.4). Sacubitril/valsartan must not be started until 36 hours after taking the last dose of perindopril therapy. Perindopril therapy must not be started until 36 hours after the last dose of sacubitril/valsartan (see sections 4.3 and 4.4). Concomitant use of ACE inhibitors with racecadotril, mTOR inhibitors (e.g. sirolimus, everolimus, temsirolimus) and gliptins (e.g. linagliptin, saxagliptin, sitagliptin, vildagliptin) may lead to an increased risk for angioedema (see section 4.4).
Drugs inducing hyperkalaemia
Although serum potassium usually remains within normal limits, hyperkalaemia may occur in some patients treated with Triveram. Some drugs or therapeutic classes may increase the occurrence of hyperkalaemia: aliskiren, potassium salts, potassium-sparing diuretics (e.g. spironolactone, triamterene or amiloride), ACE inhibitors, angiotensin-II receptors antagonists, NSAIDs, heparins, immunosuppressant agents such as ciclosporin or tacrolimus, trimethoprim and cotrimoxazole (trimethoprim/sulfamethoxazole), as trimethoprim is known to act as a potassium-sparing diuretic like amiloride. The combination of these drugs increases the risk of hyperkalaemia. Therefore, the combination of Triveram with the above-mentioned drugs is not recommended. If concomitant use is indicated, they should be used with caution and with frequent monitoring of serum potassium.
Concomitant use contraindicated (see section 4.3)
Concomitant use not recommended (see section 4.4)

Concomitant use which requires special care



Concomitant use to be taken into consideration
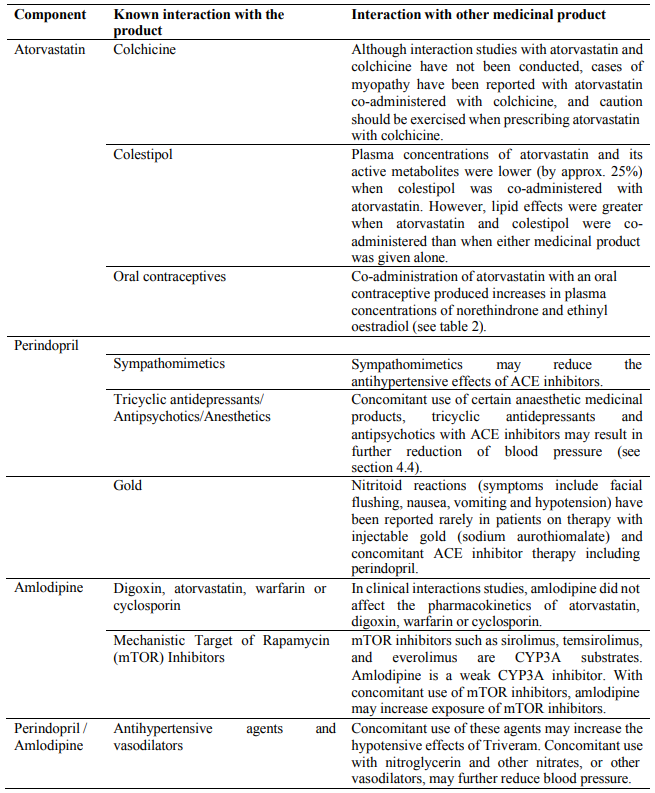
Table 1. Effect of co-administered medicinal product on the pharmacokinetics of atorvastatin:
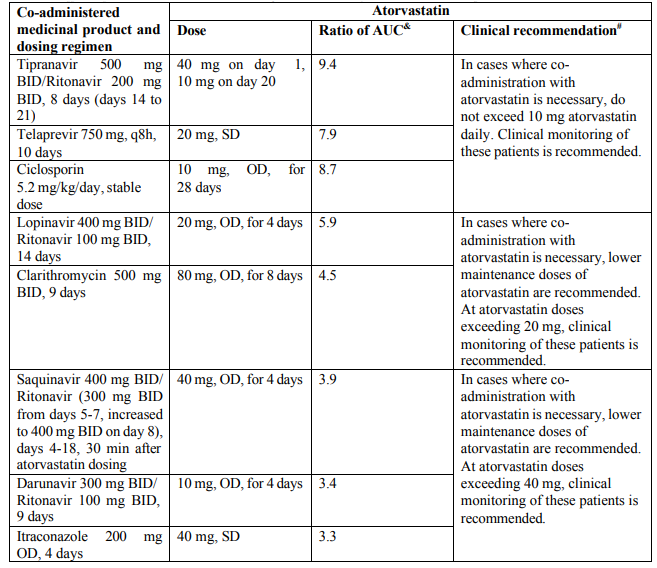
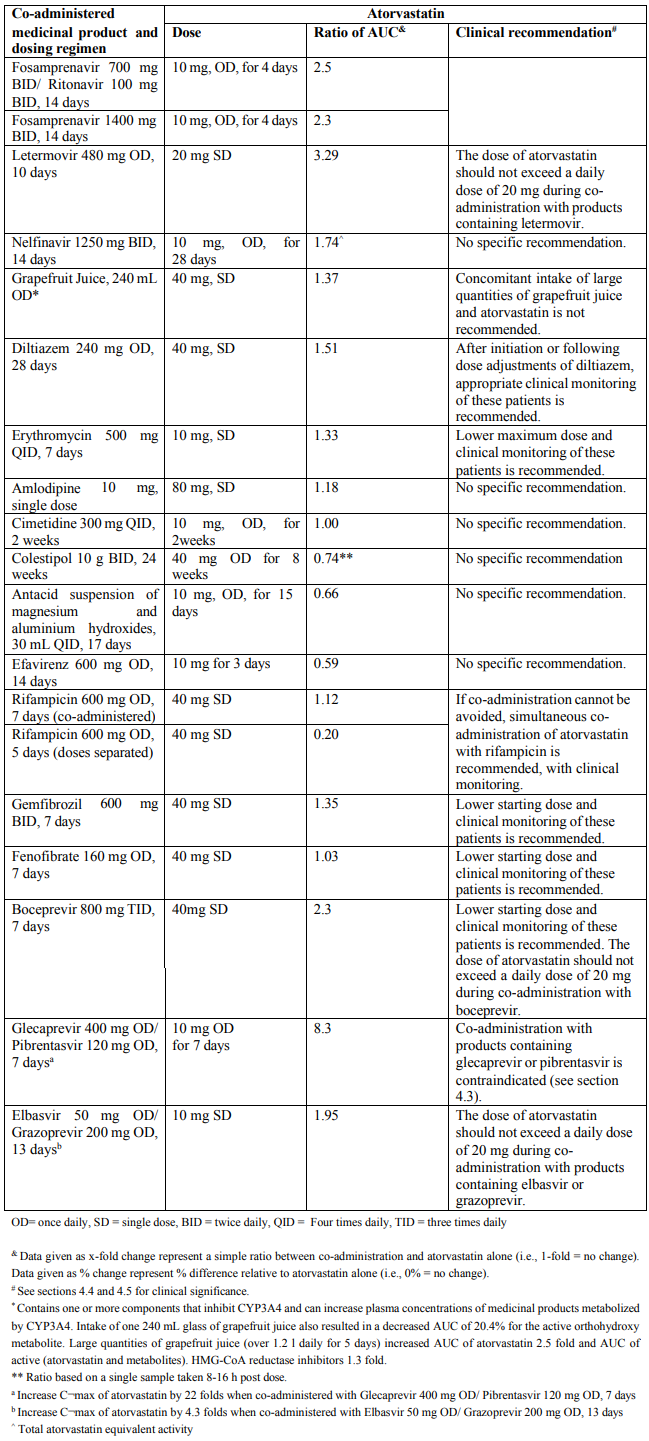
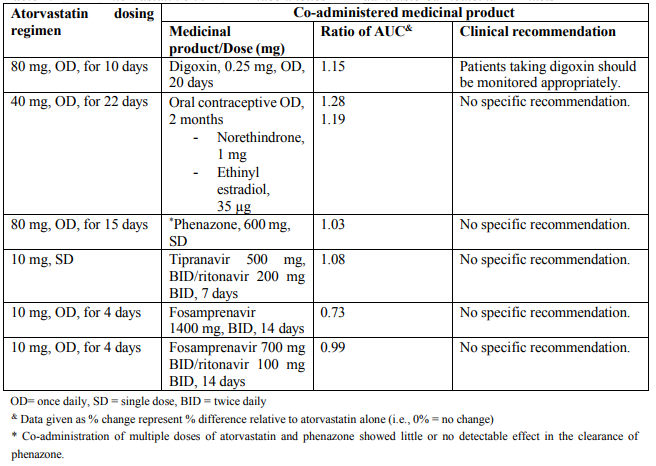
Table 2. Effect of atorvastatin on the pharmacokinetics of co-administered medicinal products:
4.6. Fertility, pregnancy and lactation
Triveram is contraindicated during pregnancy and lactation (see section 4.3).
Women of childbearing potential
Women of child-bearing potential should use appropriate contraceptive measures during treatment with Triveram (see section 4.3).
Pregnancy
Atorvastatin
Safety in pregnant women has not been established. No controlled clinical trials with atorvastatin have been conducted in pregnant women. Rare reports of congenital anomalies following intrauterine exposure to HMGCoA reductase inhibitors have been received. Animal studies have shown toxicity to reproduction (see section 5.3).
Maternal treatment with atorvastatin may reduce the fetal levels of mevalonate which is a precursor of cholesterol biosynthesis. Atherosclerosis is a chronic process, and ordinarily discontinuation of lipid-lowering medicinal products during pregnancy should have little impact on the long-term risk associated with primary hypercholesterolaemia.
For these reasons, atorvastatin should not be used in women who are pregnant, trying to become pregnant or suspected to be pregnant. Treatment with atorvastatin should be suspended for the duration of pregnancy or until confirmation of the absence of pregnancy (see section 4.3).
Perindopril
The use of ACE inhibitors is not recommended during the first trimester of pregnancy. The use of ACE inhibitors is contra-indicated during the 2nd and 3rd trimesters of pregnancy (see section 4.3).
Epidemiological evidence regarding the risk of teratogenicity following exposure to ACE inhibitors during the first trimester of pregnancy has not been conclusive; however a small increase in risk cannot be excluded. Patients planning pregnancy should be changed to alternative anti-hypertensive treatments which have an established safety profile for use in pregnancy. When pregnancy is diagnosed, treatment with ACE inhibitors should be stopped immediately, and, if appropriate, alternative therapy should be started.
Exposure to ACE inhibitor therapy during the second and third trimestersis known to induce human foetoxicity (decreased renal function, oligohydramnios, skull ossification retardation) and neonatal toxicity (renal failure, hypotension, hyperkalaemia) (see section 5.3). Should exposure to ACE inhibitor have occurred from the second trimester of pregnancy, ultrasound check of renal function and skull is recommended. Infants whose mothers have taken ACE inhibitors should be closely observed for hypotension (see also sections 4.3 and 4.4).
Amlodipine
Safety of amlodipine in human pregnancy has not been established. In animal studies, reproductive toxicity was observed at high doses (see section 5.3).
Breast-feeding
Atorvastatin
It is not known whether atorvastatin or its metabolites are excreted in human milk. In rats, plasma concentrations of atorvastatin and its active metabolites are similar to those in milk (see section 5.3). Because of the potential for serious adverse reactions, women taking atorvastatin should not breast-feed their infants. Atorvastatin is contraindicated during breastfeeding (see section 4.3).
Perindopril
Because no information is available regarding the use of perindopril during breastfeeding, perindopril is not recommended and alternative treatments with better established safety profiles during breast-feeding are preferable, especially while nursing a newborn or preterm infant.
Amlodipine
Amlodipine is excreted in human milk. The proportion of the maternal dose received by the infant has been estimated with an interquartile range of 3–7%, with a maximum of 15%. The effect of amlodipine on infants is unknown.
Fertility
Atorvastatin
In animal studies atorvastatin had no effect on male or female fertility (see section 5.3).
Perindopril
There was no effect on reproductive performance or fertility.
Amlodipine
Reversible biochemical changes in the head of spermatozoa have been reported in some patients treated by calcium channel blockers. Clinical data are insufficient regarding the potential effect of amlodipine on fertility. In one rat study, adverse effects were found on male fertility (see section 5.3).
4.7. Effects on ability to drive and use machines
No studies have been performed on the effect of Triveram on the ability to drive and use machines.
- Atorvastatin has negligible influence on the ability to drive and use machines. Perindopril has no direct influence on the ability to drive and use machines but individual reactions related to low blood pressure may occur in some patients, particularly at the start of treatment or in combination with another antihypertensive medication.
- Amlodipine can have minor or moderate influence on the ability to drive and use machines. If patients taking amlodipine suffer from dizziness, headache, fatigue or nausea the ability to react may be impaired.
As a result the ability to drive or operate machinery may be impaired in patients taking Triveram. Caution is recommended especially at the start of treatment.
4.8. Undesirable effects
Summary of the profile
The most commonly reported adverse reactions with atorvastatin, perindopril and amlodipine given separately include: nasopharyngitis, allergic reactions, hyperglycaemia, headache, pharyngolaryngeal pain, epistaxis, constipation, flatulence, dyspepsia, nausea, diarrhoea, change of bowel habit, myalgia, arthralgia, pain in extremity, muscle spasms, joint swelling, ankle swelling, back pain, liver function test abnormal, blood creatine kinase increased, somnolence, dizziness, palpitations, flushing, abdominal pain, oedema, fatigue, paresthaesia, visual disturbance, diplopia, tinnitus, vertigo, hypotension, cough, dyspnoea, vomiting, dysgeusia, rash, pruritus, asthenia.
Tabulated list of adverse reactions
The following undesirable effects have been observed during treatment with atorvastatin, perindopril, amlodipine, or given separately and ranked under the MedDRA classification by body system and under heading of frequency using the following convention: Very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from the available data)).
| MedDRA System Organ Class | Undesirable effects | Frequency | ||
|---|---|---|---|---|
| Atorvastatin | Perindopril | Amlodipine | ||
| Infections and infestation | Nasopharyngitis | Common | - | - |
| Rhinitis | - | Very rare | Uncommon | |
| Blood and lymphatic system disorders | Thrombocytopenia | Rare | Very rare | Very rare |
| Leucopenia/neutropenia | - | Very rare | Very rare | |
| Eosinophilia | - | Uncommon* | - | |
| Agranulocytosis or pancytopenia | - | Very rare | - | |
| Haemolytic anaemia in patients with a congenital deficiency of G-6PDH | - | Very rare | - | |
| Immune system disorders | Allergic reactions | Common | - | Very rare |
| Anaphylaxis | Very rare | - | - | |
| Endocrine disorders | Syndrome of inappropriate antidiuretic hormone secretion (SIADH) | - | Rare | - |
| Metabolism and nutrition disorders | Hyperglycaemia | Common | - | Very rare |
| Hypoglycaemia | Uncommon | Uncommon* | - | |
| Hyponatraemia | - | Uncommon* | - | |
| Hyperkalaemia reversible on discontinuation (see section 4.4) | - | Uncommon* | - | |
| Anorexia | Uncommon | - | - | |
| Psychiatric disorders | Insomnia | Uncommon | - | Uncommon |
| Mood swings | - | Uncommon | Uncommon | |
| Sleep disorder | Uncommon | - | ||
| Depression | - | Uncommon* | Uncommon | |
| Nightmares | Uncommon | - | - | |
| Confusion | - | Very rare | Rare | |
| Nervous system disorders | Somnolence | - | Uncommon* | Common |
| Dizziness | Uncommon | Common | Common | |
| Headache | Common | Common | Common | |
| Tremor | - | - | Uncommon | |
| Dysgeusia | Uncommon | Common | Uncommon | |
| Syncope | - | Uncommon* | Uncommon | |
| Hypoesthesia | Uncommon | - | Uncommon | |
| Paresthaesia | Uncommon | Common | Uncommon | |
| Hypertonia | - | - | Very rare | |
| Peripheral neuropathy | Rare | - | Very rare | |
| Stroke possible secondary to excessive hypotension in highrisk patients (see section 4.4) | - | Very rare | - | |
| Amnesia | Uncommon | - | - | |
| Extrapyramidal disorder (extrapyramidal syndrome) | - | - | Not known | |
| Myasthenia gravis | Not known | - | - | |
| Eye disorders | Visual disturbances | Rare | Common | Common |
| Diplopia | - | - | Common | |
| Vision blurred | Uncommon | - | - | |
| Ocular myasthenia | Not known | - | - | |
| Ear and labyrinth disorders | Tinnitus | Uncommon | Common | Uncommon |
| Vertigo | - | Common | - | |
| Hearing loss | Very Rare | - | - | |
| Cardiac disorders | Myocardial infarction secondary to excessive hypotension in high-risk patients (see section 4.4) | - | Very rare | Very rare |
| Angina pectoris (see section 4.4) | - | Very rare | - | |
| Arrhythmia (including bradycardia, ventricular tachycardia and atrial fibrillation) | - | Very rare | Uncommon | |
| Tachycardia | - | Uncommon* | - | |
| Palpitations | - | Uncommon* | Common | |
| Vascular disorders | Hypotension (and effects related to hypotension) | - | Common | Uncommon |
| Vasculitis | - | Uncommon* | Very rare | |
| Stroke possible secondary to excessive hypotension in high- risk patients (see section 4.4) | - | Very rare | - | |
| Flushing | - | Rare* | Common | |
| Raynaud’s phenomenon | - | Not known | - | |
| Respiratory, thoracic and mediastinal disorders | Pharyngolaryngeal pain | Common | - | - |
| Epistaxis | Common | - | - | |
| Cough | - | Common | Uncommon | |
| Dyspnoea | - | Common | Common | |
| Bronchospasm | - | Uncommon | - | |
| Eosinophilic pneumonia | - | Very rare | - | |
| Gastro-intestinal disorders | Nausea | Common | Common | Common |
| Vomiting | Uncommon | Common | Uncommon | |
| Abdominal pain upper and lower | Uncommon | Common | Common | |
| Dyspepsia | Common | Common | Common | |
| Diarrhoea | Common | Common | - | |
| Constipation | Common | Common | - | |
| Dry mouth | - | Uncommon | Uncommon | |
| Pancreatitis | Uncommon | Very rare | Very rare | |
| Gastritis | - | - | Very rare | |
| Gingival hyperplasia | - | - | Very rare | |
| Change of bowel habit | - | - | Common | |
| Eructation | Uncommon | - | - | |
| Flatulence | Common | - | - | |
| Hepato-biliary disorders | Hepatitis either cytolytic or cholestatic (see section 4.4) | Uncommon | Very rare | Very rare |
| Jaundice | - | - | Very rare | |
| Cholestasis | Rare | - | - | |
| Hepatic failure | Very rare | - | - | |
| Skin and subcutaneous tissue disorders | Rash | Uncommon | Common | Uncommon |
| Pruritus | Uncommon | Common | Uncommon | |
| Urticaria | Uncommon | Uncommon | Very rare | |
| Purpura | - | - | Uncommon | |
| Skin discolouration | - | - | Uncommon | |
| Hyperhidrosis | - | Uncommon | Uncommon | |
| Exanthema | - | - | Uncommon | |
| Alopecia | Uncommon | - | Uncommon | |
| Angioedema (see section 4.4) | Rare | Uncommon | Very rare | |
| Exfoliative dermatitis | - | - | Very rare | |
| Pemphigoid | - | Uncommon* | - | |
| Psoriasis aggravation | - | Rare* | - | |
| Stevens-Johnson syndrome | Rare | - | Very rare | |
| Photosensitivity reactions | - | Uncommon* | Very rare | |
| Toxic epidermal necrolysis | Rare | - | Not known | |
| Erythema multiforme | Rare | Very rare | Very rare | |
| Musculoskeletal and connective tissue disorders | Joint swelling | Common | - | - |
| Ankle swelling | - | - | Common | |
| Pain in extremity | Common | - | - | |
| Arthralgia | Common | Uncommon* | Uncommon | |
| Muscle spasms | Common | Common | Common | |
| Myalgia | Common | Uncommon* | Uncommon | |
| Back pain | Common | - | Uncommon | |
| Neck pain | Uncommon | - | - | |
| Muscle fatigue | Uncommon | - | - | |
| Myopathy | Rare | - | - | |
| Myositis | Rare | - | - | |
| Rhabdomyolysis | Rare | - | - | |
| Muscle rupture | Rare | - | - | |
| Tendinopathy sometimes complicated by rupture | Rare | - | - | |
| Lupus-like syndrome | Very rare | - | - | |
| Immune-mediated necrotizing myopathy (see section 4.4) | Not known | - | - | |
| Renal and urinary disorders | Micturition disorder | - | - | Uncommon |
| Nocturia | - | - | Uncommon | |
| Increased urinary frequency | - | - | Uncommon | |
| Renal insufficiency | - | Uncommon | - | |
| Acute renal failure | - | Rare | - | |
| Anuria/Oliguria | - | Rare* | - | |
| Reproductive system and breast disorders | Impotence/erectile dysfunction | - | Uncommon | Uncommon |
| Gynaecomastia | Very rare | - | Uncommon | |
| General disorders and administration site conditions | Asthenia | Uncommon | Common | Uncommon |
| Fatigue | Uncommon | - | Common | |
| Chest pain | Uncommon | Uncommon* | Uncommon | |
| Pain | - | - | Uncommon | |
| Malaise | Uncommon | Uncommon* | Uncommon | |
| Oedema peripheral | Uncommon | Uncommon* | Common | |
| Pyrexia | Uncommon | Uncommon* | - | |
| Investigations | Blood urea increased | - | Uncommon* | - |
| Blood creatinine increased | - | Uncommon* | - | |
| Hepatic enzymes increased | - | Rare | Very rare** | |
| Blood bilirubin increased | - | Rare | - | |
| Weight increase | Uncommon | - | Uncommon | |
| White blood cells urine positive | Uncommon | - | - | |
| Weight decrease | - | - | Uncommon | |
| Liver function test abnormal | Common | - | - | |
| Blood creatine kinase increased | Common | - | - | |
| Haemoglobin decreased and haematocrit decreased | - | Very rare | - | |
| Injury, poisoning and procedural complications | Fall | - | Uncommon* | - |
* Frequency calculated from clinical trials for adverse events detected from spontaneous report
** Mostly consistent with cholestasis
Exceptional cases of extrapyramidal syndrome have been reported with amlodipine.
As with other HMG-CoA reductase inhibitors elevated serum transaminases have been reported in patients receiving atorvastatin. These changes were usually mild, transient, and did not require interruption of treatment. Clinically important (>3 times upper normal limit) elevations in serum transaminases occurred in 0.8% patients on atorvastatin. These elevations were dose related and were reversible in all patients.
Elevated serum creatine kinase (CK) levels greater than 3 times upper limit of normal occurred in 2.5% of patients on atorvastatin, similar to other HMG-CoA reductase inhibitors in clinical trials. Levels above 10 times the normal upper range occurred in 0.4% atorvastatin -treated patients (see section 4.4).
The following adverse events have been reported with some statins:
- Sexual dysfunction.
- Depression.
- Exceptional cases of interstitial lung disease, especially with long term therapy (see section 4.4).
- Diabetes Mellitus: Frequency will depend on the presence or absence of risk factors (fasting blood glucose ≥5.6 mmol/L, BMI>30kg/m², raised triglycerides, history of hypertension).
Cognitive impairment
There have been rare post-marketing reports of cognitive impariment (e.g. memory loss, forgetfulness, amnesia, memory impairment, confusion) associated with statin use. These cognitive issues have been reported for all statins. The reports are generally non-serious, and reversible upon statin discontinuation, with variable times to symptom onset (1 day to years) and symptom resolution (median of 3 weeks).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system.
6.2. Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.