TRUQAP Film-coated tablet Ref.[111589] Active ingredients: Capivasertib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: AstraZeneca AB, SE-151 85 Södertälje, Sweden
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, other antineoplastic agents
ATC code: L01EX27
Mechanism of action
Capivasertib is a potent, selective inhibitor of the kinase activity of all 3 isoforms of serine/threonine kinase AKT (AKT1, AKT2 and AKT3). AKT is a pivotal node in the phosphatidylinositol 3-kinase (PI3K) signalling cascade regulating multiple cellular processes including cellular survival, proliferation, cell cycle, metabolism, gene transcription and cell migration. AKT activation in tumours is a result of upstream activation from other signalling pathways, mutations of AKT1, loss of Phosphatase and Tensin Homolog (PTEN) function and mutations in the catalytic subunit of PI3K (PIK3CA).
Capivasertib reduces growth of cell lines derived from solid tumours and haematological disease, including breast cancer cell lines with and without PIK3CA or AKT1 mutations, or PTEN alterations.
Treatment with capivasertib and fulvestrant demonstrated anti-tumour response in a range of ER+ human breast cancer PDX models representative of different breast cancer subsets. This included models with and without detectable mutations or alterations in PIK3CA, PTEN or AKT1.
Cardiac electrophysiology
Based on an exposure-response analysis of data from 180 patients with advanced solid malignancies who received capivasertib doses from 80 to 800 mg, the predicted QTcF prolongation was 3.87 ms at the mean steady state Cmax following 400 mg twice daily.
Clinical efficacy
CAPItello-291 was a randomised, double-blind, placebo-controlled study that enrolled 708 patients, designed to study the efficacy and safety of TRUQAP in combination with fulvestrant in adult females, pre- or post-menopausal, and adult males with locally advanced (inoperable) or metastatic ER-positive and HER2-negative (defined as IHC 0 or 1+, or IHC 2+/ISH-) breast cancer of which 289 patients had tumors with one or more eligible PIK3CA/AKT1/PTEN alterations following recurrence or progression on or after aromatase inhibitor (AI)-based treatment.
Patients were excluded if they had more than 2 lines of endocrine therapy for locally advanced (inoperable) or metastatic disease, more than 1 line of chemotherapy for locally advanced (inoperable) or metastatic disease, prior treatment with AKT, PI3K, mTOR inhibitors, fulvestrant and/or other SERDs, clinically significant abnormalities of glucose metabolism (defined as patients with diabetes mellitus Type 1 or Type 2 requiring insulin treatment, and/or HbA1c ˃8.0% (63.9 mmol/mol)), history of clinically significant cardiac disease, and symptomatic visceral disease or any disease burden that makes the patient ineligible for endocrine therapy.
Patients were randomised 1:1 to receive either 400 mg of TRUQAP (N=355) or placebo (N=353) given twice daily for 4 days followed by 3 days off treatment each week of 28-day treatment cycle. Fulvestrant 500 mg was administered on cycle 1 days 1 and 15 and then at day 1 of a 28-day cycle. Pre- or perimenopausal women were treated with an LHRH agonist. Randomisation was stratified by presence of liver metastases, prior treatment with CDK4/6 inhibitors and geographical region. Treatment was administered until disease progression, death, withdrawal of consent, or unacceptable toxicity. A tumour sample was collected prior to randomisation to determine PIK3CA/AKT1/PTEN alteration status retrospectively by central testing.
Demographic and baseline characteristics were well balanced between arms. Of the 708 patients, the median age was 58 years (range 26 to 90 and 30.7% were over 65 years of age); female (99%); White (57.5%), Asian (26.7%), Black (1.1%); Eastern Cooperative Oncology Group (ECOG) performance status 0 (65.7%), 1 (34.2%), 21.8% were pre- or perimenopausal. All patients received prior endocrine-based therapy (100% AI-based treatment and 44.1% received tamoxifen). Prior treatment with CDK4/6 inhibitor was reported in 70.1% of patients. Chemotherapy for locally advanced (inoperable) or metastatic disease was reported in 18.2% of patients. Patient demographics for those in the PIK3CA/AKT1/PTEN-altered subgroup were generally representative of the overall study population.
The dual primary endpoints were investigator assessed progression free survival (PFS) in the overall population and PFS in the PIK3CA/AKT1/PTEN-altered subgroup per Response Evaluation Criteria in Solid Tumours (RECIST) v1.1.
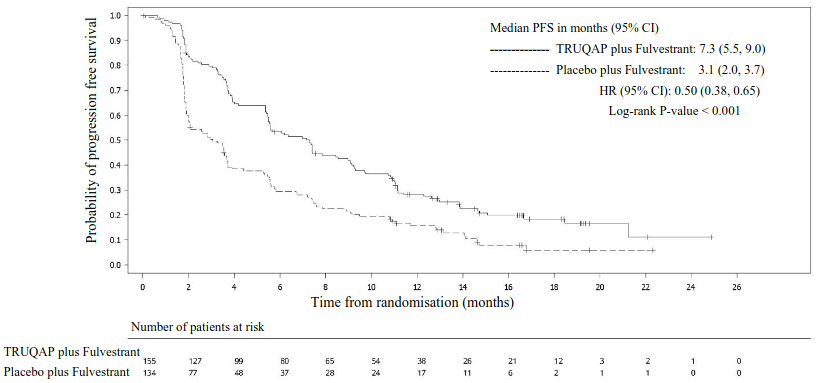
At the data cutoff date (DCO) of 15 August 2022, the study showed statistically significant improvement in PFS for patients receiving TRUQAP plus fulvestrant compared to patients receiving placebo plus fulvestrant, in both the overall population and the PIK3CA/AKT1/PTEN-altered subgroup (see table 9). An exploratory analysis of PFS in the 313 (44%) patients whose tumours did not have a PIK3CA/AKT1/PTEN alterations showed a HR of 0.79 (95% CI: 0.61, 1.02), indicating that the difference in the overall population was primarily attributed to the results seen in the population of patients whose tumours have PIK3CA/AKT1/PTEN alteration. PFS results by investigator assessment were supported by consistent results from a blinded independent central review (BICR) assessment. OS data were immature at the time of the primary PFS analysis. The investigator-assessed ORR in patients receiving TRUQAP plus fulvestrant and placebo plus fulvestrant was 22.9% and 12.2%, respectively, in the overall population and 28.8% and 9.7%, respectively, in the altered subgroup.
Efficacy results are presented in Table 9 and Figure 1.
Table 9. Progression-free survival, by investigator assessment in PIK3CA/AKT1/PTEN-altered subgroup:
| PIK3CA/AKT1/PTEN-altered subgroup N=289 | ||
|---|---|---|
| TRUQAP plus fulvestrant N=155 | Placebo plus fulvestrant N=134 | |
| Number of PFS events – n (%) | 121 (78.1) | 115 (85.8) |
| Median PFS months (95% CI) | 7.3 (5.5, 9.0) | 3.1 (2.0, 3.7) |
| Hazard ratio (95% CI)a | 0.50 (0.38, 0.65) | |
| p-valueb | <0.001 | |
a Stratified Cox proportional hazards model. A hazard ratio <1 favours capivasertib + fulvestrant. For the Overall population, log-rank test and Cox model stratified by presence of liver metastases (yes vs no), prior use of CDK4/6 inhibitors (yes vs no) and geographic region (Region 1: United States, Canada, Western Europe, Australia, and Israel, Region 2: Latin America, Eastern Europe and Russia vs Region 3: Asia). For the altered population, the log rank test and Cox model stratified by presence of liver metastases (yes vs no), and prior use of CDK4/6 inhibitors (yes vs no).
b Stratified log-rank test.
Figure 1. Kaplan-Meier plot of progression-free survival – CAPItello-291 (investigator assessment, PIK3CA/AKT1/PTEN-altered subgroup):
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with TRUQAP in all subsets of the paediatric population in breast cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Capivasertib pharmacokinetics have been characterised in healthy subjects and patients with solid tumours. The systemic exposure (AUC and Cmax) increased proportionally over the dose range of 80 to 800 mg range after single dose administration in patients. After multiple-dose administration of 80 to 600 mg twice daily, the AUC increased slightly more than dose proportional. Following intermittent dosing of capivasertib 400 mg twice daily, 4 days on and 3 days off, the capivasertib steady-state concentrations with AUC of 8 069 hng/mL (37%) and Cmax of 1 371 ng/mL (30%) are predicted to be attained on the 3rd and 4th dosing day of each week, starting from week 2. During the off-dosing days, the plasma concentrations are low (approximately 0.5% to 15% of the steady state Cmax).
Absorption
Capivasertib is rapidly absorbed with peak concentration (Cmax) observed at approximately 1-2 hours in patients. The mean absolute bioavailability is 29%.
Food effect
When capivasertib was administered after a high-fat, high-calorie meal (approximately 1000 kcal), the fed to fasted ratio was 1.32 and 1.23, for AUC and Cmax, respectively, compared to when given after an overnight fast. When capivasertib was administered after a low-fat, low-calorie meal (approximately 400 kcal), the exposure was similar to that after fasted administration with fed to fasted ratios of 1.14 and 1.21, for AUC and Cmax, respectively. Co-administration with food did not result in clinically relevant changes to the exposure.
Distribution
The mean volume of distribution was 2.6 L/Kg after intravenous administration to healthy subjects. Capivasertib is not extensively bound to plasma protein (percentage unbound 22%) and the plasma to blood ratio is 0.71.
Biotransformation
Capivasertib is primarily metabolised by CYP3A4 and UGT2B7 enzymes. The major metabolite in human plasma was an ether glucuronide that accounted for 83% of total drug-related material. A minor oxidative metabolite was quantified at 2% and capivasertib accounted for 15% of total circulating drug-related material. No active metabolites have been identified.
Elimination
The effective half-life after multiple dosing in patients was 8.3 h. The mean total plasma clearance was 38 L/h after a single IV administration to healthy subjects. The mean total oral plasma clearance was 60 L/h after single oral administration and decreased by 8% after repeated dosing of 400 mg twice daily. Following single oral dose of 400 mg, the mean total recovery of radioactive dose was 45% from urine and 50% from faeces. Renal clearance was 21% of total clearance. Capivasertib is primarily eliminated by metabolism.
Special populations
Effect of race, age, gender and weight
Based on population pharmacokinetic analysis, AUC and Cmax showed that race (including White and Japanese patients), gender or age did not significantly impact the capivasertib exposure. There was a statistically significant correlation of apparent oral clearance of capivasertib to body weight. Compared to a patient with a body weight of 66 kg, a 47 kg patient is predicted to have 12% higher AUC. There is no basis for dose modification based on body weight as the predicted effect on capivasertib exposure was small.
Renal impairment
Based on population pharmacokinetic analyses, AUC and Cmax were 1% higher in patients with mild renal impairment (creatinine clearance 60 to 89 mL/min), compared to patients with normal renal 18 function. AUC and Cmax were 16% higher in patients with moderate renal impairment (creatinine clearance 30 to 59 mL/min), compared to patients with normal renal function. There is no data in severe renal impairment or end-stage renal disease (creatinine clearance <30 ml/min).
Hepatic impairment
Based on population pharmacokinetic analyses, AUC and Cmax were 5% higher in patients with mild hepatic impairment (bilirubin ≤ ULN and AST > ULN, or bilirubin > 1 ULN to ≤ 1.5 ULN), compared to patients with normal hepatic function (bilirubin ≤ ULN and AST ≤ ULN). AUC was 17% and Cmax was 13% higher in patients with moderate hepatic impairment (bilirubin > 1.5 ULN to ≤ 3 ULN), compared to patients with normal hepatic function. There is limited data in patients with moderate hepatic impairment and no data in severe hepatic impairment.
Drug-drug interactions
Co-administration of a single dose of capivasertib 400 mg after repeated dosing of acid-reducing agent rabeprazole 20 mg BID for 3 days in healthy subjects did not result in clinically relevant changes of the capivasertib exposure.
In vitro studies have demonstrated that capivasertib is primarily metabolised by CYP3A4 and UGT2B7 enzymes. Results of clinical drug-drug interaction (DDI) studies investigating potential DDI based on CYP3A4 interactions (itraconazole and enzalutamide) are included in section 4.5 above. Clinical DDI studies investigating potential DDIs based on UGT2B7 interactions have not been performed.
Capivasertib inhibited CYP2C9, CYP2D6, CYP3A4 and UGT1A1 and induced CYP1A2, CYP2B6 and CYP3A4 metabolising enzymes in in vitro studies. It also inhibited BCRP, OATP1B1, OATP1B3, OAT3, OCT2, MATE1 and MATE2K drug transporters in vitro. Results of clinical DDI study investigating potential DDIs based on CYP3A4 interactions (midazolam) are included in section 4.5 above. Clinical DDI studies investigating potential DDIs based on CYP1A2, CYP2B6, CYP2C9, CYP2D6, UGT1A1, BCRP, OATP1B1, OATP1B3, OAT3, OCT2, MATE1 and MATE2K interactions have not been performed.
5.3. Preclinical safety data
Non-clinical/Repeat-dose toxicity
The major target organs or systems for toxicity were insulin signalling (increased levels of glucose and insulin in rats and dogs), the male reproductive organs (tubular degeneration in rats and dogs), and the renal system in rats (polyuria, decreased tubular epithelial cell size, decreased kidney size and weight). The findings present following 1 month of dosing were largely reversible within 1 month of cessation of dosing. Findings occurred at plasma concentrations lower or similar to those in humans (approximately 0.14 to 2 times) at the recommended dose of 400 mg twice daily (based on total AUC). Cardiovascular effects (QTc interval prolongation, increased cardiac contractility, and decreased blood pressure) were seen in dogs at plasma concentrations approximately 1.4 to 2.7 times the expected clinical exposure in humans at the recommended dose of 400 mg twice daily (based on unbound Cmax).
Mutagenicity and carcinogenicity
Capivasertib showed no mutagenic or genotoxic potential in vitro. When dosed orally to rats, capivasertib induced micronuclei in the bone marrow via an aneugenic mode of action.
Carcinogenicity studies have not been conducted with capivasertib.
Reproductive toxicity
Embryofoetal/Developmental toxicity
In a rat embryofoetal study, capivasertib caused an increase in post implantation loss, an increase in early embryonic deaths, together with reduced gravid uterine and foetal weights, and minor foetal visceral variations. These effects were seen at a dose level of 150 mg/kg/day which caused maternal toxicity, and where plasma concentrations were approximately 0.8 times the exposure in humans at the recommended dose of 400 mg twice daily (based on total AUC). When capivasertib was administered to pregnant rats at 150 mg/kg/day throughout gestation and through early lactation, there was a reduction in litter and pup weights.
Exposure to capivasertib was confirmed in suckling pups which may indicate the potential for excretion of capivasertib in human milk.
Fertility
Capivasertib has resulted in testicular toxicity and may impair fertility in males of reproductive potential. Effects on female fertility have not been studied in animals. In females, repeat-dose toxicity studies have reported some weight changes of the uterus in rats which were attributed to estrous cycle changes. Histopathological examination conducted in rat and dog studies did not show any treatment-related effects on female reproductive organs, which may be indicative of an adverse effect on female fertility.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.