VANFLYTA Film-coated tablet Ref.[51535] Active ingredients: Quizartinib
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Daiichi Sankyo Europe GmbH, Zielstattstrasse 48, 81379 Munich, Germany
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors
ATC code: L01EX11
Mechanism of action
Quizartinib is an inhibitor of the receptor tyrosine kinase FLT3. Quizartinib and its major metabolite AC886 competitively bind to the adenosine triphosphate (ATP) binding pocket of FLT3 with high affinity. Quizartinib and AC886 inhibit FLT3 kinase activity, preventing autophosphorylation of the receptor, thereby inhibiting further downstream FLT3 receptor signalling and blocking FLT3-ITDdependent cell proliferation.
Pharmacodynamic effects
Cardiac electrophysiology
The exposure-response analysis of QuANTUM-First predicted a concentration-dependent QTcF interval prolongation of 24.1 ms [upper bound of two-sided 90% confidence interval (CI): 26.6 ms] at the steady-state Cmax of quizartinib (53 mg) during maintenance therapy.
Clinical efficacy and safety
The efficacy and safety of quizartinib vs. placebo was investigated in a randomised, double-blind, placebo-controlled, phase III study, QuANTUM-First. The study enrolled 539 adult patients between 18 and 75 years of age (25% were 65 years or older), who were newly diagnosed with FLT3-ITD positive AML, as determined prospectively by a clinical study assay. Patients were randomised (1:1) to receive VANFLYTA 35.4 mg once daily (n=268) or placebo (n=271) for two weeks in each cycle in combination with standard chemotherapy (induction followed by consolidation for responding patients) followed by single-agent maintenance therapy with VANFLYTA (26.5 mg once daily for two weeks and 53 mg once daily thereafter) or placebo for up to 36 cycles (28 days/cycle).
Patients received up to 2 cycles of induction chemotherapy with either daunorubicin on days 1, 2 and 3 or idarubicin on days 1, 2 and 3 and cytarabine for 7 days, followed by post remission therapy which consisted of up to 4 cycles of consolidation chemotherapy and/or HSCT. Consolidation chemotherapy consisted of cytarabine on days 1, 3 and 5. Patients who proceeded to HSCT stopped receiving study treatment 7 days before the start of a conditioning regimen. Please refer to the Summary of Product Characteristics for daunorubicin, idarubicin and cytarabine dosing recommendations.
The two randomised treatment groups were well balanced with respect to baseline demographics, disease characteristics and stratification factors. Of the 539 patients, the median age was 56 years (range 20-75 years), 26.1% of patients in the quizartinib arm and 24% of patients in the placebo arm were 65 years or older; 54.5% were female and 45.5% were male; 59.7% were White, 29.3% were Asian, 1.3% were Black or African American, and 9.7% were other races. Eighty-four percent of patients had an Eastern Cooperative Oncology Group (ECOG) baseline performance status of 0 or 1. The majority of the patients (72.4%) had intermediate cytogenetics risk status at baseline. FLT3-ITD variant allele frequency (VAF) was 3-25% in 35.6% of patients, greater than 25-50% in 52.1% of patients and greater than 50% in 12.1% of patients.
The primary efficacy measure was overall survival (OS) defined as the time from randomisation until death from any cause.
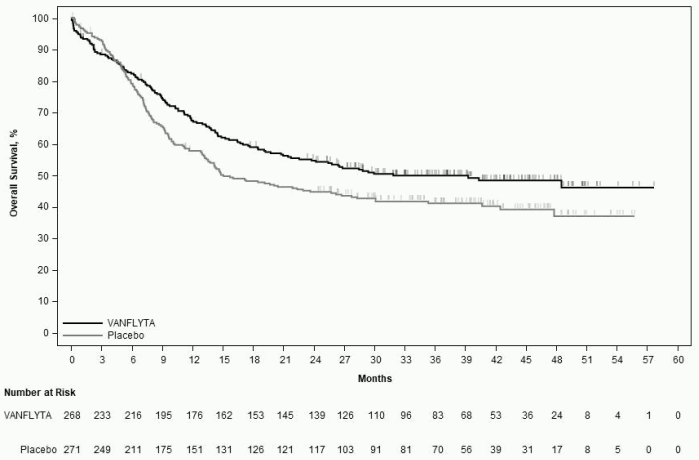
The study demonstrated a statistically significant improvement in OS for the quizartinib arm (see Table 5 and Figure 1). The median follow-up time of the study was 39.2 months.
A difference was observed between the quizartinib arm vs. the placebo arm in the estimates of survival rates (95% CI) at the landmark timepoints of 12, 24, 36 and 48 months (see Table 5).
The complete remission (CR) rate [95% CI] for quizartinib was 54.9% (147/268) [48.7, 60.9] vs. 55.4% (150/271) [49.2, 61.4] for placebo.
Table 5. Efficacy results from QuANTUM-First (intent-to-treat population):
| Quizartinib N=268 | Placebo N=271 | |
|---|---|---|
| OS (months) | ||
| Median (95% CI)a | 31.9 (21.0, NE) | 15.1 (13.2, 26.2) |
| HRb relative to placebo (95% CI) | 0.776 (0.615, 0.979) | |
| p-value (two-sided stratified log-rank test) | 0.0324 | |
| OS rate () (95 CI)a | ||
| 12 months | 67.4 (61.3, 72.7) | 57.7 (51.6, 63.4) |
| 24 months | 54.7 (48.4, 60.5) | 44.7 (38.7, 50.6) |
| 36 months | 49.9 (43.7, 55.9) | 41.1 (35.0, 47.0) |
| 48 months | 48.4 (41.9, 54.5) | 37.0 (29.8, 44.2) |
CI = confidence interval; NE = not estimable
a Kaplan-Meier estimate
b Hazard ratio (HR) was based on stratified Cox regression model.
Figure 1. Kaplan-Meier curves for overall survival in QuANTUM-First:
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with VANFLYTA in one or more subsets of the paediatric population in the treatment of acute myeloid leukaemia (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics of quizartinib and its active metabolite AC886, were evaluated in healthy adult subjects (single dose) and in patients with newly diagnosed AML (steady state).
Absorption
The absolute bioavailability of quizartinib from the tablet formulation was 71%. After oral administration under fasted conditions in healthy subjects, time to peak concentration (median tmax) of quizartinib and AC886 measured post dose was approximately 4 hours (range 2 to 8 hours) and 5 to 6 hours (range 4 to 120 hours), respectively.
The administration of quizartinib with food, in healthy subjects, decreased quizartinib Cmax by 1.09-fold, increased AUCinf by 1.08-fold and tmax was delayed by two hours. These changes in exposure are not considered clinically relevant. VANFLYTA can be administered with or without food.
Based on population pharmacokinetic modelling in newly diagnosed AML patients, at 35.4 mg/day, steady state during induction therapy, the geometric mean (CV) Cmax of quizartinib and AC886 was estimated to be 140 ng/mL (71) and 163 ng/mL (52%), respectively, and the geometric mean (CV) AUC0-24h was 2 680 ng•h/mL (85) and 3 590 ng•h/mL (51%), respectively.
During consolidation therapy at 35.4 mg/day, steady state, the geometric mean (CV) Cmax of quizartinib and AC886 was estimated to be 204 ng/mL (64) and 172 ng/mL (47%), respectively, and the geometric mean (CV) AUC0-24h was 3 930 ng•h/mL (78) and 3 800 ng•h/mL (46%), respectively.
During maintenance therapy at 53 mg/day, steady state, the geometric mean (CV) Cmax of quizartinib and AC886 was estimated to be 529 ng/mL (60) and 262 ng/mL (48%), respectively, and the geometric mean (CV) AUC0-24h was 10 200 ng•h/mL (75) and 5 790 ng•h/mL (46%), respectively.
Distribution
In vitro binding of quizartinib and AC886 to human plasma proteins is greater than or equal to 99%.
The blood-to-plasma ratio of quizartinib and AC886 are concentration dependent, indicating saturation of the distribution to erythrocytes. At clinically relevant plasma concentrations, the blood-to-plasma ratio is approximately 1.3 for quizartinib and approximately 2.8 for AC886. Blood-to-plasma ratio of AC886 is also dependent on haematocrit, with a trend of increasing at higher haematocrit levels.
The geometric mean (CV) volume of distribution of quizartinib in healthy subjects was estimated to be 275 L (17).
Biotransformation
Quizartinib is primarily metabolised by CYP3A4 and CYP3A5 in vitro via oxidative pathways which produces the active metabolite AC886, which is then further metabolised by CYP3A4 and CYP3A5. The steady-state AC886-to-quizartinib AUC0-24h ratio during maintenance therapy was 0.57.
Elimination
The mean (SD) effective half-lives (t1/2) for quizartinib and AC886 are 81 hours (73) and 136 hours (113), respectively, in patients with newly diagnosed AML. The mean (SD) accumulation ratios (AUC0-24h) for quizartinib and AC886 were 5.4 (4.4) and 8.7 (6.8), respectively.
Quizartinib and its metabolites are primarily eliminated by the hepatobiliary route with excretion mainly via faeces (76.3% of the orally administered radioactive dose). Unchanged quizartinib represented approximately 4% of the orally administered radioactive dose in faeces. Renal excretion is a minor route of elimination of the administered radioactive dose (<2%).
The geometric mean (CV) total body clearance (CL) of quizartinib in healthy subjects was estimated to be 2.23 L/hour (29).
Linearity/non-linearity
Quizartinib and AC886 showed linear kinetics in the dose range of 26.5 mg to 79.5 mg in healthy subjects and 17.7 mg to 53 mg in AML patients.
Pharmacokinetic/pharmacodynamic relationships
Age (18 to 91 years), race, sex, body weight, or renal impairment (CLcr 30 to 89 mL/min, estimated by Cockcroft-Gault) did not have a clinically relevant effect on quizartinib and AC886 exposure based on a population pharmacokinetic analysis.
Interaction studies with other medicinal products
Transporters
In vitro studies showed that quizartinib is a substrate for P-gp but not for BCRP, OATP1B1, OATP1B3, OCT1, OAT2, MATE1 or MRP2. AC886 is a substrate for BCRP but not for OATP1B1, OATP1B3, MATE1 or MRP2. However, the single-dose administration of quizartinib with ketoconazole, a strong inhibitor for both CYP3A and P-gp, increased quizartinib Cmax by approximately 1.17-fold, suggesting that the effect of P-gp is minimal. As dose adjustment is required for concomitant strong CYP3A inhibitors, many of which also inhibit P-gp, no specific dose adjustment is required for P-gp inhibitors.
Uridine diphosphate glucuronosyltransferases (UGT)1A1 substrates
Quizartinib inhibits UGT1A1 with an estimated in vitro Ki of 0.78 μM. Based on a physiologically based pharmacokinetic (PBPK) analysis, quizartinib was predicted to increase the Cmax and AUCinf of raltegravir (a UGT1A1 substrate) by 1.03-fold which was not considered clinically relevant.
Special populations
Hepatic impairment
In a single-dose (26.5 mg) phase 1 study, the pharmacokinetics of quizartinib and AC886 were assessed in subjects with mild hepatic impairment (Child-Pugh Class A) or moderate hepatic impairment (Child-Pugh Class B ) and compared to subjects with normal hepatic function. The exposure (Cmax and AUCinf) of quizartinib and AC886 were similar (≤30% difference) across all groups. Protein binding of quizartinib and AC886 is not affected by impaired hepatic function. Therefore, hepatic impairment did not have a clinically relevant effect on quizartinib and AC886 exposure.
No dose adjustment is recommended in patients with mild or moderate hepatic impairment.
Patients with severe hepatic impairment (Child-Pugh Class C) were not included in the clinical studies; therefore, VANFLYTA is not recommended for use in these patients.
Renal impairment
A population pharmacokinetic analysis in AML patients with mild to moderate renal impairment (CLcr 30 to 89 mL/min) showed that renal function did not affect quizartinib and AC886 clearance. Therefore, mild and moderate renal impairment did not have a clinically relevant effect on quizartinib and AC886 exposure. No dose adjustment is recommended in patients with mild or moderate renal impairment.
Patients with severe renal impairment (CLcr <30 mL/min) were not included in the clinical studies; therefore, VANFLYTA is not recommended for use in these patients.
5.3. Preclinical safety data
In genotoxicity studies, quizartinib was mutagenic in a bacterial reverse mutation assay, but not in a mammalian cell mutation assay (mouse lymphoma thymidine kinase) or in an in vivo transgenic rodent mutation assay. Quizartinib was not clastogenic and did not induce polyploidy in a chromosome aberration assay and was not clastogenic or aneugenic in a single-dose rat bone marrow micronucleus assay. An in vivo bone marrow micronucleus assay in rats was equivocal after 28 days repeated dosing. After a single higher dose, the result was negative.
Fertility studies in animals have not been conducted with quizartinib. However, adverse findings in male and female reproductive systems were observed in repeat dose toxicity studies in rats and monkeys. In female rats, ovarian cysts and vaginal mucosal modifications were observed at doses approximately 10 times the recommended human dose (RHD) based on AUC. Findings in female monkeys included atrophy of the uterus, ovary and vagina; observed at doses approximately 0.3 times the RHD based on AUC. The corresponding no observed adverse effect levels (NOAELs) for these changes were 1.5 times and 0.1 times the RHD, respectively, based on AUC. In male rats, testicular seminiferous tubular degeneration and failure of sperm release were observed at approximately 8 times the RHD based on AUC. Findings in male monkeys included germ cell depletion in the testes; observed at approximately 0.5 times the RHD based on AUC. The corresponding NOAELs for these changes were 1.4 times and 0.1 times the RHD, respectively, based on AUC. After a four-week recovery period, all these findings except the vaginal mucosal modifications in the female rats were reversible.
In embryo-foetal toxicity studies, embryo-foetal lethality and increased post-implantation loss were observed at maternally toxic doses. Foetotoxicity (lower foetal weights, effects on skeletal ossification) and teratogenicity (foetal abnormalities including oedema) were observed at doses approximately 3 times the RHD based on AUC. The NOAEL was 0.5 times the RHD based on AUC. Quizartinib is considered to be potentially teratogenic.
Animal toxicology studies
In repeat dose toxicity studies, haematopoietic and lymphoid organ toxicity were observed including decreased peripheral blood cells and bone marrow hypocellularity; liver toxicity including elevated aminotransferases, hepatocellular necrosis and birefringent crystal deposition (dogs); and kidney toxicity including tubular basophilia and birefringent crystal deposition (male rats). These changes were noted at approximately 0.4 times, 0.4 times and 9 times the RHD based on AUC, respectively. The corresponding NOAELs were approximately 0.1 times, 0.1 times and 1.5 times the RHD based on AUC, respectively.
In vitro and animal safety pharmacology studies
In cardiovascular safety pharmacology studies conducted in cynomolgus monkeys, quizartinib resulted in QT prolongation at doses approximately 2 times the RHD of 53 mg/day based on Cmax. The NOAEL was approximately 0.4 times the RHD based on Cmax. Quizartinib primarily inhibited IKs with a maximum inhibition of 67.5% at 2.9 µM. The maximum inhibition of IKs by AC886 was 26.9% at 2.9 µM. Quizartinib and AC886 at 3 μM statistically significantly inhibited hERG currents by 16.4% and 12.0%, respectively. Neither quizartinib nor AC886 inhibited INa, INa-L and ICa-L at any concentration tested.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.