VENCLYXTO Film-coated tablet Ref.[9093] Active ingredients: Venetoclax
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: AbbVie Deutschland GmbH & Co. KG, Knollstrasse, 67061, Ludwigshafen, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, other antineoplastic agents
ATC code: L01XX52
Mechanism of action
Venetoclax is a potent, selective inhibitor of B-cell lymphoma (BCL)-2, an anti-apoptotic protein. Overexpression of BCL-2 has been demonstrated in CLL and AML cells where it mediates tumour cell survival and has been associated with resistance to chemotherapeutics. Venetoclax binds directly to the BH3-binding groove of BCL-2, displacing BH3 motif-containing pro-apoptotic proteins like BIM, to initiate mitochondrial outer membrane permeabilization (MOMP), caspase activation, and programmed cell death. In non-clinical studies, venetoclax has demonstrated cytotoxic activity in tumour cells that overexpress BCL-2.
Pharmacodynamic effects
Cardiac electrophysiology
The effect of multiple doses of venetoclax up to 1200 mg once daily on the QTc interval was evaluated in an open-label, single-arm study in 176 patients. Venetoclax had no effect on QTc interval and there was no relationship between venetoclax exposure and change in QTc interval.
Clinical efficacy and safety
Chronic lymphocytic leukaemia
Venetoclax in combination with obinutuzumab for the treatment of patients with previously untreated CLL – study BO25323 (CLL14)
A randomised (1:1), multicentre, open-label phase 3 study evaluated the efficacy and safety of venetoclax + obinutuzumab versus obinutuzumab + chlorambucil in patients with previously untreated CLL and comorbidities (total Cumulative Illness Rating Scale [CIRS] score >6 or creatinine clearance [CrCl] <70 ml/min). Patients in the study were assessed for risk of TLS and received prophylaxis accordingly prior to obinutuzumab administration. All patients received obinutuzumab at 100 mg on Cycle 1 Day 1, followed by 900 mg which could have been administered on Day 1 or Day 2, then 1000 mg doses on Days 8 and 15 of Cycle 1, and on Day 1 of each subsequent cycle, for a total of 6 cycles. On Day 22 of Cycle 1, patients in the venetoclax + obinutuzumab arm began the 5-week venetoclax dose-titration schedule, continuing through Cycle 2 Day 28. Upon completion of the dose- titration schedule, patients continued venetoclax 400 mg once daily from Cycle 3 Day 1 until the last day of Cycle 12. Each cycle was 28 days. Patients randomised to the obinutuzumab + chlorambucil arm received 0.5 mg/kg oral chlorambucil on Day 1 and Day 15 of Cycles 1-12. Patients continued to be followed for disease progression and overall survival (OS) after completing therapy.
Baseline demographic and disease characteristics were similar between the study arms. The median age was 72 years (range: 41 to 89 years), 89% were white, and 67% were male; 36% and 43% were Binet stage B and C, respectively. The median CIRS score was 8.0 (range: 0 to 28) and 58% of patients had CrCl <70 ml/min. A 17p deletion was detected in 8% of patients, TP53 mutations in 10%, 11q deletion in 19%, and unmutated IgVH in 57%. The median follow-up at the time of the primary analysis was 28 months (range: 0 to 36 months).
At baseline, the median lymphocyte count was 55 x 109 cells/l in both study arms. On Cycle 1 Day 15, the median count had decreased to 1.03 x 109 cells/l (range: 0.2 to 43.4 x 109 cells/l) in the obinutuzumab + chlorambucil arm and 1.27 x 109 cells/l (range: 0.2 to 83.7 x 109 cells/l) in the venetoclax + obinutuzumab arm.
Progression-free survival (PFS) was assessed by investigators using the International Workshop for Chronic Lymphocytic Leukemia (IWCLL) updated National Cancer Institute-sponsored Working Group (NCI-WG) guidelines (2008).
At the time of the primary analysis (data cut-off date 17 August 2018), 14% (30/216) of patients in the venetoclax + obinutuzumab arm had a PFS event of disease progression or death compared with 36% (77/216) in the obinutuzumab + chlorambucil arm, as assessed by investigators (hazard ratio [HR]: 0.35 [95% confidence interval [CI]: 0.23, 0.53]; p<0.0001, stratified log-rank test). Median PFS was not reached in either study arm.
Progression-free-survival was also assessed by an Independent Review Committee (IRC) and was consistent with the investigator-assessed PFS.
Investigator-assessed overall response rate (ORR) was 85% (95% CI: 79.2, 89.2) and 71% (95% CI: 64.8, 77.2) in the venetoclax + obinutuzumab and obinutuzumab + chlorambucil arms, respectively (p=0.0007, Cochran-Mantel-Haenszel test). Investigator-assessed complete remission + complete remission with incomplete marrow recovery (CR + CRi) rate was 50% and 23% in the venetoclax + obinutuzumab and obinutuzumab + chlorambucil arms, respectively (p<0.0001, Cochran-Mantel-Haenszel test).
Minimal residual disease (MRD) at the end of treatment was evaluated using allele-specific oligonucleotide polymerase chain reaction (ASO-PCR) assay. MRD negativity was defined as less than one CLL cell per 104 leukocytes. MRD negativity rates in peripheral blood were 76% (95% CI: 69.2, 81.1) in the venetoclax + obinutuzumab arm compared to 35% (95% CI: 28.8, 42.0) in the obinutuzumab + chlorambucil arm (p<0.0001). Per protocol, MRD in bone marrow was to be assessed only in responding patients (CR/CRi and partial remission [PR]). MRD negativity rates in the bone marrow were 57% (95% CI: 50.1, 63.6) in the venetoclax + obinutuzumab arm and 17% (95% CI: 12.4, 22.8) in the obinutuzumab + chlorambucil arm (p<0.0001).
65-month follow-up:
Efficacy was assessed after a median follow-up of 65 months (data cut-off date 8 November 2021). Efficacy results for the CLL14 65-month follow-up are presented in Table 10. The Kaplan-Meier curve of investigator-assessed PFS is shown in Figure 1.
Table 10. Investigator-assessed efficacy results in CLL14 (65-month follow-up):
| Endpoint | Venetoclax + obinutuzumab N=216 | Obinutuzumab + chlorambucil N=216 |
|---|---|---|
| Progression-free survival | ||
| Number of events (%) | 80 (37) | 150 (69) |
| Median, months (95% CI) | NR (64.8, NE) | 36.4 (34.1, 41.0) |
| Hazard ratio, stratified (95% CI) | 0.35 (0.26, 0.46) | |
| Overall survival | ||
| Number of events (%) | 40 (19) | 57 (26) |
| Hazard ratio, stratified (95% CI) | 0.72 (0.48, 1.09) | |
CI = confidence interval; NE = not evaluable; NR = not reached
Figure 1. Kaplan-Meier curve of investigator-assessed progression-free survival (intent-to-treat population) in CLL14 with 65-month follow-up:

The PFS benefit with venetoclax + obinutuzumab versus obinutuzumab + chlorambucil treatment was observed across all subgroups of patients evaluated, including high-risk patients with deletion 17p and/or TP53 mutation and/or unmutated IgVH.
Venetoclax in combination with rituximab for the treatment of patients with CLL who have received at least one prior therapy – study GO28667 (MURANO)
A randomised (1:1), multicentre, open-label phase 3 study evaluated the efficacy and safety of venetoclax + rituximab versus bendamustine + rituximab in patients with previously treated CLL. Patients in the venetoclax + rituximab arm completed the Venclyxto 5-week dose-titration schedule and then received 400 mg once daily for 24 months from Cycle 1 Day 1 of rituximab in the absence of disease progression or unacceptable toxicity. Rituximab was initiated after the 5-week dose-titration schedule at 375 mg/m² for Cycle 1 and 500 mg/m² for Cycles 2-6. Each cycle was 28 days. Patients randomised to bendamustine + rituximab received bendamustine at 70 mg/m² on Days 1 and 2 for 6 cycles and rituximab as described above.
Median age was 65 years (range: 22 to 85); 74% were male, and 97% were white. Median time since diagnosis was 6.7 years (range: 0.3 to 29.5). Median prior lines of therapy was 1 (range: 1 to 5); and included alkylating agents (94%), anti-CD20 antibodies (77%), B-cell receptor pathway inhibitors (2%) and prior purine analogues (81%, including 55% fludarabine + cyclophosphamide + rituximab (FCR)). At baseline, 47% of patients had one or more nodes ≥5 cm, and 68% had ALC ≥25 x 109/l. A 17p deletion was detected in 27% of patients, TP53 mutations in 26%, 11q deletion in 37%, and unmutated IgVH gene in 68%. Median follow-up time for primary analysis was 23.8 months (range: 0.0 to 37.4 months).
Progression-free survival was assessed by investigators using the IWCLL updated NCI-WG guidelines (2008).
At the time of the primary analysis (data cut-off date 8 May 2017), 16% (32/194) of patients in the venetoclax + rituximab arm had experienced a PFS event, compared with 58% (114/195) in the bendamustine + rituximab arm (HR: 0.17 [95% CI: 0.11, 0.25]; p<0.0001, stratified log-rank test). The PFS events included 21 disease progression and 11 death events in the venetoclax + rituximab arm, and 98 disease progression and 16 death events in the bendamustine + rituximab arm. Median PFS 23 was not reached in the venetoclax + rituximab arm and was 17.0 months (95% CI: 15.5, 21.6) in the bendamustine + rituximab arm.
The 12- and 24-month PFS estimates were 93% (95% CI: 89.1, 96.4) and 85% (95% CI: 79.1, 90.6) in the venetoclax + rituximab arm and 73% (95% CI: 65.9, 79.1) and 36% (95% CI: 28.5, 44.0) in the bendamustine + rituximab arm, respectively.
Efficacy results for the primary analysis were also assessed by an IRC demonstrating a statistically significant 81% reduction in the risk of progression or death for patients treated with venetoclax + rituximab (HR: 0.19 [95% CI: 0.13, 0.28]; p<0.0001).
Investigator-assessed ORR for patients treated with venetoclax + rituximab was 93% (95% CI: 88.8, 96.4), with a CR + CRi rate of 27%, nodular partial remission (nPR) rate of 3%, and PR rate of 63%. For patients treated with bendamustine + rituximab, ORR was 68% (95% CI: 60.6, 74.2), with a CR + CRi rate of 8%, nPR rate of 6%, and PR rate of 53%. Median duration of response (DOR) was not reached with median follow-up of approximately 23.8 months. The IRC-assessed ORR for patients treated with venetoclax + rituximab was 92% (95% CI: 87.6, 95.6), with a CR + CRi rate of 8%, nPR rate of 2%, and PR rate of 82%. For patients treated with bendamustine + rituximab, IRC-assessed ORR was 72% (95% CI: 65.5, 78.5), with a CR + CRi rate of 4%, nPR rate of 1%, and PR rate of 68%. The discrepancy between IRC- and investigator-assessed CR rates was due to interpretation of residual adenopathy on CT scans. Eighteen patients in the venetoclax + rituximab arm and 3 patients in the bendamustine + rituximab arm had negative bone marrow and lymph nodes <2 cm.
MRD at the end of combination treatment was evaluated using ASO-PCR and/or flow cytometry. MRD negativity was defined as less than one CLL cell per 104 leukocytes. MRD negativity rates in peripheral blood were 62% (95% CI: 55.2, 69.2) in the venetoclax + rituximab arm compared to 13% (95% CI: 8.9, 18.9) in the bendamustine + rituximab arm. Of those with MRD assay results available in peripheral blood, 72% (121/167) in the venetoclax + rituximab arm and 20% (26/128) in the bendamustine + rituximab arm were found to be MRD negative. MRD negativity rates in the bone marrow were 16% (95% CI: 10.7, 21.3) in the venetoclax + rituximab arm and 1% (95% CI: 0.1, 3.7) in the bendamustine + rituximab arm. Of those with MRD assay results available in bone marrow, 77% (30/39) in the venetoclax + rituximab arm and 7% (2/30) in the bendamustine + rituximab arm were found to be MRD negative.
Median OS had not been reached in either treatment arm. Death occurred in 8% (15/194) of patients treated with venetoclax + rituximab and 14% (27/195) of patients treated with bendamustine + rituximab (hazard ratio: 0.48 [95% CI: 0.25, 0.90]).
By the data cut-off date, 12% (23/194) of patients in the venetoclax + rituximab arm and 43% (83/195) of patients in the bendamustine + rituximab arm had started a new anti-leukaemic treatment or died (stratified hazard ratio: 0.19; [95% CI: 0.12, 0.31]). The median time to new anti-leukaemic treatment or death was not reached in the venetoclax + rituximab arm and was 26.4 months in the bendamustine + rituximab arm.
59-month follow-up:
Efficacy was assessed after a median follow-up of 59 months (data cut-off date 8 May 2020). Efficacy results for the MURANO 59-month follow-up are presented in Table 11.
Table 11. Investigator-assessed efficacy results in MURANO (59-month follow-up):
| Endpoint | Venetoclax + rituximab N=194 | Bendamustine + rituximab N=195 |
|---|---|---|
| Progression-free survival | ||
| Number of events (%)a | 101 (52) | 167 (86) |
| Median, months (95% CI) | 54 (48.4, 57.0) | 17 (15.5, 21.7) |
| Hazard ratio, stratified (95% CI) | 0.19 (0.15, 0.26) | |
| Overall survival | ||
| Number of events (%) | 32 (16) | 64 (33) |
| Hazard ratio (95% CI) | 0.40 (0.26, 0.62) | |
| 60-month estimate, % (95% CI) | 82 (76.4, 87.8) 62 (54.8, 69.6) | |
| Time to next anti-leukaemic treatment | ||
| Number of events (%)b | 89 (46) | 149 (76) |
| Median, months (95% CI) | 58 (55.1, NE) | 24 (20.7, 29.5) |
| Hazard ratio, stratified (95% CI) | 0.26 (0.20, 0.35) | |
| MRD negativityc | ||
| Peripheral blood at end of treatment, n (%)d | 83 (64) | NAf |
| 3-year PFS estimate from end of treatment, % (95% CI)e | 61 (47.3, 75.2) | NAf |
| 3-year OS estimate from end of treatment, % (95% CI)e | 95 (90.0, 100.0) | NAf |
CI = confidence interval; MRD = minimal residual disease; NE = not evaluable; OS = overall survival; PFS = progression-free survival; NA = not applicable.
a 87 and 14 events in the venetoclax + rituximab arm were due to disease progression and death, compared to 148 and 19 events in the bendamustine + rituximab arm, respectively.
b 68 and 21 events in the venetoclax + rituximab arm were due to patients starting a new anti-leukaemic treatment and death, compared to 123 and 26 events in the bendamustine + rituximab arm, respectively.
c Minimal residual disease was evaluated using allele-specific oligonucleotide polymerase chain reaction (ASO-PCR) and/or flow cytometry. The cut-off for a negative status was one CLL cell per 104 leukocytes.
d In patients who completed venetoclax treatment without progression (130 patients).
e In patients who completed venetoclax treatment without progression and were MRD negative (83 patients).
f No equivalent to end of treatment visit in bendamustine + rituximab arm.
In total, 130 patients in the venetoclax + rituximab arm completed 2 years of venetoclax treatment without progression. For these patients, the 3-year PFS estimate post-treatment was 51% (95% CI: 40.2, 61.9).
The Kaplan-Meier curve of investigator-assessed PFS is shown in Figure 2.
Figure 2. Kaplan-Meier curve of investigator-assessed progression-free survival (intent-to-treat population) in MURANO (data cut-off date 8 May 2020) with 59-month follow-up:

Results of subgroup analyses:
The observed PFS benefit of venetoclax + rituximab compared with bendamustine + rituximab was consistently observed across all subgroups of patients evaluated, including high-risk patients with deletion 17p/TP53 mutation and/or unmutated IgVH (Figure 3).
Figure 3. Forest plot of investigator-assessed progression-free survival in subgroups from MURANO (data cut-off date 8 May 2020) with 59-month follow-up:

17p deletion status was determined based on central laboratory test results.
Final overall survival analysis (86-month follow-up):
At the time of the final OS analysis (data cut-off date 03 August 2022), a total of 144 randomised patients had died; 60/194 patients (31%) in the venetoclax + rituximab arm and 84/195 patients (43%) in the bendamustine + rituximab arm. The median OS was not reached in the venetoclax + rituximab arm and was 88 months in the bendamustine + rituximab arm. The estimated risk of death was decreased by 47% for patients treated with venetoclax + rituximab (stratified HR = 0.53; 95% CI: 0.37, 0.74). The final OS analysis was not type I error controlled. The Kaplan-Meier curve of overall survival is shown in Figure 4.
Figure 4. Kaplan-Meier curve of overall survival (intent-to-treat population) in MURANO (data cut-off date 03 August 2022) with 86-month follow-up:

Venetoclax as monotherapy for the treatment of patients with CLL harbouring 17p deletion or TP53 mutation – study M13-982
The safety and efficacy of venetoclax in 107 patients with previously treated CLL with 17p deletion were evaluated in a single-arm, open-label, multicentre study (M13-982). Patients followed a 4- to 5-week dose-titration schedule starting at 20 mg and increasing to 50 mg, 100 mg, 200 mg and finally 400 mg once daily. Patients continued to receive venetoclax 400 mg once daily until disease progression or unacceptable toxicity was observed. The median age was 67 years (range: 37 to 85 years); 65% were male, and 97% were white. The median time since diagnosis was 6.8 years (range: 0.1 to 32 years; N=106). The median number of prior anti-CLL treatments was 2 (range: 1 to 10 treatments); 49.5% with a prior nucleoside analogue, 38% with prior rituximab, and 94% with a prior alkylator (including 33% with prior bendamustine). At baseline, 53% of patients had one or more nodes ≥5 cm, and 51% had ALC ≥25 x 109/l. Of the patients, 37% (34/91) were fludarabine refractory, 81% (30/37) harboured the unmutated IgVH gene, and 72% (60/83) had TP53 mutation. The median time on treatment at the time of evaluation was 12 months (range: 0 to 22 months).
The primary efficacy endpoint was ORR as assessed by an IRC using the IWCLL updated NCI-WG guidelines (2008). Efficacy results are shown in Table 12. Efficacy data are presented for 107 patients with data cut-off date 30 April 2015. An additional 51 patients were enrolled in a safety expansion cohort. Investigator-assessed efficacy results are presented for 158 patients with a later data cut-off date 10 June 2016. The median time on treatment for 158 patients was 17 months (range: 0 to 34 months).
Table 12. Efficacy results in patients with previously treated CLL with 17p deletion (study M13-982):
| Endpoint | IRC assessment (N=107)a | Investigator assessment (N=158)b |
|---|---|---|
| Data cutoff date | 30 April 2015 | 10 June 2016 |
| ORR, % (95% CI) | 79 (70.5, 86.6) | 77 (69.9, 83.5) |
| CR + CRi, % | 7 | 18 |
| nPR, % | 3 | 6 |
| PR, % | 69 | 53 |
| DOR, months, median (95% CI) | NR | 27.5 (26.5, NR) |
| PFS, % (95% CI) 12-month estimate 24-month estimate | 72 (61.8, 79.8) NA | 77 (69.1, 82.6) 52 (43, 61) |
| PFS, months, median (95% CI) | NR | 27.2 (21.9, NR) |
| TTR, months, median (range) | 0.8 (0.1-8.1) | 1.0 (0.5-4.4) |
a One patient did not harbour the 17p deletion.
b Includes 51 additional patients from the safety expansion cohort.
CI = confidence interval; CR = complete remission; CRi = complete remission with incomplete marrow recovery; DOR = duration of response; IRC = independent review committee; nPR = nodular PR; NA = not available; NR = not reached; ORR = overall response rate; PFS = progression-free survival, PR = partial remission; TTR = time to first response.
Minimal residual disease (MRD) was evaluated using flow cytometry in 93 of 158 patients who achieved CR, CRi, or PR with limited remaining disease with venetoclax treatment. MRD negativity was defined as a result below 0.0001 (<1 CLL cell per 104 leukocytes in the sample). Twenty-seven percent (42/158) of patients were MRD negative in the peripheral blood, including 16 patients who were also MRD negative in the bone marrow.
Venetoclax as monotherapy for the treatment of patients with CLL who have failed a B-cell receptor pathway inhibitor – study M14-032
The efficacy and safety of venetoclax in patients with CLL who had been previously treated with and failed ibrutinib or idelalisib therapy were evaluated in an open-label, multicentre, non-randomised, phase 2 study (M14-032). Patients received venetoclax via a recommended dose-titration schedule. Patients continued to receive venetoclax 400 mg once daily until disease progression or unacceptable toxicity was observed.
At the time of data cut-off (26 July 2017), 127 patients were enrolled and treated with venetoclax. Of these, 91 patients had received prior ibrutinib therapy (Arm A) and 36 had received prior idelalisib therapy (Arm B). The median age was 66 years (range: 28 to 85 years), 70% were male, and 92% were white. The median time since diagnosis was 8.3 years (range: 0.3 to 18.5 years; N=96). Chromosomal aberrations were 11q deletion (34%, 43/127), 17p deletion (40%, 50/126), TP53 mutation (38%, 26/68) and unmutated IgVH (78%, 72/92). At baseline, 41% of patients had one or more nodes ≥5 cm and 31% had ALC ≥25 x 109/l. The median number of prior oncology treatments was 4 (range: 1 to 15) in ibrutinib-treated patients and 3 (range: 1 to 11) in idelalisib-treated patients. Overall, 65% of patients received prior nucleoside analogue, 86% rituximab, 39% other monoclonal antibodies, and 72% alkylating agent (including 41% with bendamustine). At the time of evaluation, median duration of treatment with venetoclax was 14.3 months (range: 0.1 to 31.4 months).
The primary efficacy endpoint was ORR according to IWCLL updated NCI-WG guidelines. Response assessments were performed at 8 weeks, 24 weeks, and every 12 weeks thereafter.
Table 13. Efficacy results as assessed by investigator in patients who have failed a B-cell receptor pathway inhibitor (study M14-032):
| Endpoint | Arm A (ibrutinib failures) (N=91) | Arm B (idelalisib failures) (N=36) | Total (N=127) |
|---|---|---|---|
| ORR, % (95% CI) | 65 (54.1, 74.6) | 67 (49.0, 81.4) | 65 (56.4, 73.6) |
| CR + CRi, % | 10 | 11 | 10 |
| nPR, % | 3 | 0 | 2 |
| PR, % | 52 | 56 | 53 |
| PFS, % (95% CI) 12-month estimate 24-month estimate | 75 (64.7, 83.2) 51 (36.3, 63.9) | 80 (63.1, 90.1) 61 (39.6, 77.4) | 77 (68.1, 83.4) 54 (41.8, 64.6) |
| PFS, months, median (95% CI) | 25 (19.2, NR) | NR (16.4, NR) | 25 (19.6, NR) |
| OS, % (95% CI) 12-month estimate | 91 (82.8, 95.4) | 94.2 (78.6, 98.5) | 92 (85.6, 95.6) |
| TTR, months, median (range) | 2.5 (1.6-14.9) | 2.5 (1.6-8.1) | 2.5 (1.6-14.9) |
| 17p deletion and/or TP53 mutation status ORR, % (95% CI) | |||
| Yes | (n=28) 61 (45.4, 74.9) | (n=7) 58 (27.7, 84.8) | (n=35) 60 (46.6, 73.0) |
| No | (n=31) 69 (53.4, 81.8) | (n=17) 71 (48.9, 87.4) | (n=48) 70 (57.3, 80.1) |
CI = confidence interval; CR = complete remission; CRi = complete remission with incomplete marrow recovery; nPR = nodular PR; NR = not reached, ORR = overall response rate; OS = overall survival; PFS = progression-free survival, PR = partial remission, TTR = time to first response.
The efficacy data were further evaluated by an IRC demonstrating a combined ORR of 70% (Arm A: 70%; Arm B: 69%). One patient (ibrutinib failure) achieved CRi. The ORR for patients with 17p deletion and/or TP53 mutation was 72% (33/46) (95% CI: 56.5, 84.0) in Arm A and 67% (8/12) (95% CI: 34.9, 90.1) in Arm B. For patients without 17p deletion and/or TP53 mutation, the ORR was 69% (31/45) (95% CI: 53.4, 81.8) in Arm A and 71% (17/24) (95% CI: 48.9, 87.4) in Arm B.
Median OS and DOR were not reached with median follow-up of approximately 14.3 months for Arm A and 14.7 months for Arm B.
Twenty-five percent (32/127) of patients were MRD negative in the peripheral blood, including 8 patients who were also MRD negative in bone marrow.
Acute myeloid leukaemia
Venetoclax was studied in adult patients who were ≥75 years of age, or who had comorbidities that precluded the use of intensive induction chemotherapy based on at least one of the following criteria: baseline Eastern Cooperative Oncology Group (ECOG) performance status of 2–3, severe cardiac or pulmonary comorbidity, moderate hepatic impairment, creatinine clearance (CrCl) <45 ml/min, or other comorbidity.
Venetoclax in combination with azacitidine for the treatment of patients with newly diagnosed AML - study M15-656 (VIALE-A)
VIALE-A was a randomised (2:1), double-blind, placebo-controlled phase 3 study that evaluated the efficacy and safety of venetoclax in combination with azacitidine in patients with newly diagnosed AML who were ineligible for intensive chemotherapy.
Patients in VIALE-A completed the 3-day daily titration schedule to a final 400 mg once daily dose during the first 28-day cycle of treatment (see section 4.2) and received venetoclax 400 mg orally once daily thereafter in subsequent cycles. Azacitidine at 75 mg/m² was administered either intravenously or subcutaneously on Days 1-7 of each 28-day cycle beginning on Cycle 1 Day 1. During the titration, patients received TLS prophylaxis and were hospitalised for monitoring. Once bone marrow assessment confirmed a remission, defined as less than 5% leukaemia blasts with grade 4 cytopenia following Cycle 1 treatment, venetoclax or placebo was interrupted up to 14 days or until ANC ≥500/microlitre and platelet count ≥50 × 103/microlitre. For patients with resistant disease at the end of Cycle 1, a bone marrow assessment was performed after Cycle 2 or 3 and as clinically indicated. Azacitidine was resumed on the same day as venetoclax or placebo following interruption (see section 4.2). Azacitidine dose reduction was implemented in the clinical study for management of hematologic toxicity (see azacitidine Summary of Product Characteristics). Patients continued to receive treatment cycles until disease progression or unacceptable toxicity.
A total of 431 patients were randomised: 286 to the venetoclax + azacitidine arm and 145 to the placebo + azacitidine arm. Baseline demographic and disease characteristics were similar between the venetoclax + azacitidine and placebo + azacitidine arms. Overall, the median age was 76 years (range: 49 to 91 years), 76% were white, 60% were males, and ECOG performance status at baseline was 0 or 1 for 55% of patients, 2 for 40% of patients, and 3 for 5% of patients. There were 75% of patients with de novo AML and 25% with secondary AML. At baseline, 29% of patients had bone marrow blast count <30%, 22% of patients had bone marrow blast count ≥30% to <50%, and 49% had ≥50%. Intermediate or poor cytogenetic risk was present in 63% and 37% patients, respectively. The following mutations were identified: TP53 mutations in 21% (52/249), IDH1 and/or IDH2 mutation in 24% (89/372), 9% (34/372) with IDH1, 16% (58/372) with IDH2, 16% (51/314) with FLT3, and 18% (44/249) with NPM1.
The primary efficacy endpoints of the study were overall survival (OS), measured from the date of randomisation to death from any cause and composite CR rate (complete remission + complete remission with incomplete blood count recovery [CR+CRi]). The overall median follow-up at the time of analysis was 20.5 months (range: <0.1 to 30.7 months).
Venetoclax + azacitidine demonstrated a 34% reduction in the risk of death compared with placebo + azacitidine (p<0.001). Results are shown in Table 14.
Table 14. Efficacy results in VIALE-A:
| Endpoint | Venetoclax + azacitidine | Placebo + azacitidine |
|---|---|---|
| Overall survivala | (N=286) | (N=145) |
| Number of events n (%) | 161 (56) | 109 (75) |
| Median survival, months (95% CI) | 14.7 (11.9, 18.7) | 9.6 (7.4, 12.7) |
| Hazard ratiob (95% CI) | 0.66 (0.52, 0.85) | |
| p-valueb | <0.001 | |
| CR+CRi ratec | (N=147) | (N=79) |
| n (%) | 96 (65) | 20 (25) |
| (95% CI) | (57, 73) | (16, 36) |
| p-valued | <0.001 | |
CI = confidence interval; CR = (complete remission) was defined as absolute neutrophil count >1,000/microlitre, platelets >100,000/microlitre, red blood cell transfusion independence, and bone marrow with <5% blasts. Absence of circulating blasts and blasts with Auer rods; absence of extramedullary disease; CRi = complete remission with incomplete blood count recovery.
a Kaplan-Meier estimate at the second interim analysis (data cut-off date 4 January 2020).
b Hazard ratio estimate (venetoclax +azacitidine vs. placebo + azacitidine) is based on Cox-proportional hazards model stratified by cytogenetics (intermediate risk, poor risk) and age (18 to <75, ≥75) as assigned at randomisation; p-value based on log-rank test stratified by the same factors.
c The CR+CRi rate is from a planned interim analysis of first 226 patients randomised with 6 months of follow-up at the first interim analysis (data cut-off date 1 October 2018).
d P-value is from Cochran-Mantel-Haenszel test stratified by age (18 to <75, ≥75) and cytogenetic risk (intermediate risk, poor risk) as assigned at randomisation.
Figure 5. Kaplan-Meier curve for overall survival in VIALE-A:
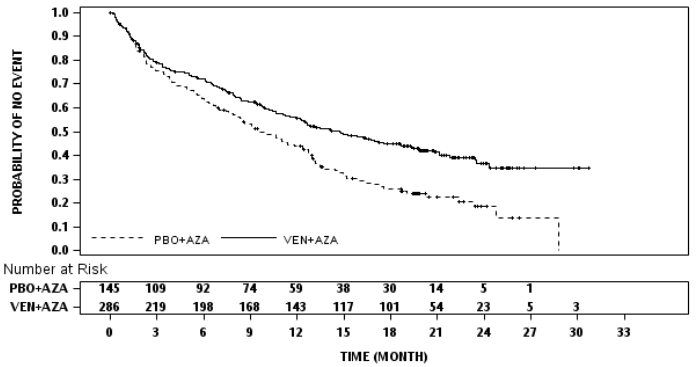
Key secondary efficacy endpoints are presented in Table 15.
Table 15. Additional efficacy endpoints in VIALE-A:
| Endpoint | Venetoclax + azacitidine N=286 | Placebo + azacitidine N=145 | |||
|---|---|---|---|---|---|
| CR rate | |||||
| n (%) | 105 (37) | 26 (18) | |||
| (95% CI) | (31, 43) | (12, 25) | p-valuea | <0.001 | |
| Median DORb, months | 17.5 | 13.3 | |||
| (95% CI) | (15.3, -) | (8.5, 17.6) | |||
| CR+CRi rate | |||||
| n (%) | 190 (66) | 41 (28) | |||
| (95% CI) | (61, 72) | (21, 36) | Median DORb, months | 17.5 | 13.4 |
| (95% CI) | (13.6, -) | (5.8, 15.5) | |||
| CR+CRi rate by initiation of Cycle 2 | |||||
| n (%) | 124 (43) | 11 (8) | |||
| (95% CI) | (38, 49) | (4, 13) | |||
| p-valuea | <0.001 | ||||
| Transfusion independence rate, platelets | |||||
| n (%) | 196 (69) | 72 (50) | |||
| (95% CI) | (63, 74) | (41, 58) | |||
| p-valuea | <0.001 | ||||
| Transfusion independence rate, red blood cells | |||||
| n (%) | 171 (60) | 51 (35) | |||
| (95% CI) | (54, 66) | (27, 44) | |||
| p-valuea | <0.001 | ||||
| CR+CRi MRD response rated | |||||
| n (%) | 67 (23) | 11 (8) | |||
| (95% CI) | (19, 29) | (4, 13) | |||
| p-valuea | <0.001 | ||||
| Event-free survival | Number of events, n (%) | 191 (67) | 122 (84) | ||
| Median EFSe, months | 9.8 | 7.0 | |||
| (95% CI) | (8.4, 11.8) | (5.6, 9.5) | |||
| Hazard ratio (95% CI)c | 0.63 (0.50, 0.80) | ||||
| p-valuec | <0.001 | ||||
CI = confidence interval; CR = complete remission; CRi = complete remission with incomplete blood count recovery; DOR = duration of response; EFS = event-free survival; MRD = minimal/measurable residual disease; n = number of responses or number of events; - = not reached.
CR (complete remission) was defined as absolute neutrophil count >1,000/microlitre, platelets >100,000/microlitre, red blood cell transfusion independence, and bone marrow with <5% blasts. Absence of circulating blasts and blasts with Auer rods; absence of extramedullary disease.
Transfusion independence was defined as a period of at least consecutive 56 days (≥56 days) with no transfusion after the first dose of study drug and on or before the last dose of the study drug + 30 days, or before relapse or disease progression or before the initiation of post treatment therapy whichever is earlier.
a P-value is from Cochran-Mantel-Haenszel test stratified by age (18 to <75, ≥75) and cytogenetic risk (intermediate risk, poor risk) as assigned at randomisation.
b DOR (duration of response) was defined as time from first response of CR for DOR of CR, from first response of CR or CRi for DOR of CR+CRi, to the first date of confirmed morphologic relapse, confirmed progressive disease or death due to disease progression, whichever occurred earlier. Median DOR is from Kaplan-Meier estimate.
c Hazard ratio estimate (venetoclax + azacitidine vs. placebo + azacitidine) is based on Cox-proportional hazards model stratified by age (18 to <75, ≥75) and cytogenetics (intermediate risk, poor risk) as assigned at randomisation; p-value based on log-rank test stratified by the same factors.
d CR+CRi MRD response rate is defined as the % of patients achieving a CR or CRi and demonstrated an MRD response of <10-3 blasts in bone marrow as determined by a standardized, central multicolour flow cytometry assay.
e Kaplan-Meier estimate.
Of patients with the FLT3 mutation, the CR+CRi rates were 72% (21/29; [95% CI: 53, 87]) and 36% (8/22; [95% CI: 17, 59]) in the venetoclax + azacitidine and placebo + azacitidine arms, respectively (p=0.021).
Of patients with IDH1/IDH2 mutations, the CR+CRi rates were 75% (46/61; [95% CI: 63, 86]) and 11% (3/28; [95% CI: 2, 28]) in the venetoclax + azacitidine and placebo + azacitidine arms, respectively (p<0.001).
Of the patients who were RBC transfusion dependent at baseline and treated with venetoclax + azacitidine, 49% (71/144) became transfusion independent. Of the patients who were platelet transfusion dependent at baseline and treated with venetoclax + azacitidine, 50% (34/68) became transfusion independent.
The median time to first response of CR or CRi was 1.3 months (range: 0.6 to 9.9 months) with venetoclax + azacitidine treatment. The median time to best response of CR or CRi was 2.3 months (range: 0.6 to 24.5 months).
Figure 6. Forest plot of overall survival by subgroups from VIALE-A:
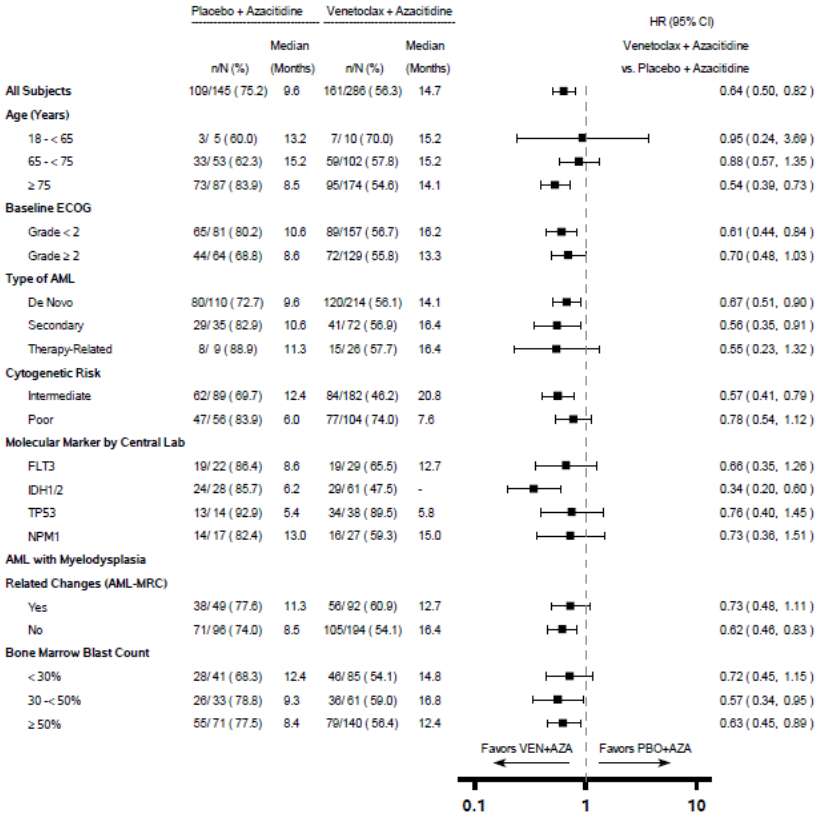
- = Not reached.
For the pre-specified secondary endpoint OS in the IDH1/2 mutation subgroup, p<0.0001 (unstratified log-rank test).
Unstratified hazard ratio (HR) is displayed on the X-axis with logarithmic scale.
Venetoclax in combination with azacitidine or decitabine for the treatment of patients with newly diagnosed AML - M14-358
Study M14-358 was a non-randomised phase ½ clinical study of venetoclax in combination with azacitidine (n=84) or decitabine (n=31) in patients with newly diagnosed AML who were ineligible for intensive chemotherapy. Patients received venetoclax via a daily titration to a final 400 mg once daily dose. The administration of azacitidine in M14-358 was similar to that of VIALE-A randomised study. Decitabine at 20 mg/m² was administered intravenously on Days 1-5 of each 28-day cycle beginning on Cycle 1 Day 1.
The median follow-up was 40.4 months (range: 0.7 to 42.7 months) for venetoclax + decitabine.
The median age of patients treated with venetoclax + decitabine was 72 years (range: 65-86 years), 87% were white, 48% males, and 87% had ECOG score 0 or 1. The CR+CRi rate was 74% (95% CI: 55, 88) in combination with decitabine.
Elderly patients
Of the 194 patients with previously treated CLL who received venetoclax in combination with rituximab, 50% were 65 years or older.
Of the 107 patients who were evaluated for efficacy from M13-982 study, 57% were 65 years or older.
Of the 127 patients who were evaluated for efficacy from M14-032 study, 58% were 65 years or older.
Of the 352 patients evaluated for safety from 3 open-label monotherapy studies, 57% were 65 years or older.
Of the 283 patients with newly diagnosed AML treated in the VIALE-A (venetoclax + azacitidine arm) clinical study, 96% were ≥65 years of age and 60% were ≥75 years of age.
Of the 31 patients treated with venetoclax in combination with decitabine in the M14-358 clinical study, 100% were ≥65 years of age and 26% were ≥75 years of age.
There were no clinically meaningful differences in safety or efficacy observed between older and younger patients in the combination and monotherapy studies.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Venclyxto in all subsets of the paediatric population in CLL (see section 4.2 for information on paediatric use).
The European Medicines Agency has deferred the obligation to submit the results of studies with Venclyxto in one or more subsets of the paediatric population in AML (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
Following multiple oral administrations, maximum plasma concentration of venetoclax was reached 5-8 hours after dose. Venetoclax steady state AUC increased proportionally over the dose range of 150-800 mg. Under low-fat meal conditions, venetoclax mean (± standard deviation) steady state Cmax was 2.1 ± 1.1 mcg /ml and AUC24 was 32.8 ± 16.9 mcg •h/ml at the 400 mg once daily dose.
Effect of food
Administration with a low-fat meal increased venetoclax exposure by approximately 3.4-fold and administration with a high-fat meal increased venetoclax exposure by 5.1- to 5.3-fold compared to fasting conditions. It is recommended that venetoclax should be administered with a meal (see section 4.2).
Distribution
Venetoclax is highly bound to human plasma protein with unbound fraction in plasma <0.01 across a concentration range of 1-30 micromolar (0.87-26 mcg/ml). The mean blood-to-plasma ratio was 0.57.
The population estimate for apparent volume of distribution (Vdss/F) of venetoclax ranged from 256-321 L in patients.
Biotransformation
In vitro studies demonstrated that venetoclax is predominantly metabolised by cytochrome P450 CYP3A4. M27 was identified as a major metabolite in plasma with an inhibitory activity against BCL-2 that is at least 58-fold lower than venetoclax in vitro.
In vitro interaction studies
Co-administration with CYP and UGT substrates:
In vitro studies indicated that venetoclax is not an inhibitor or inducer of CYP1A2, CYP2B6, CYP2C19, CYP2D6, or CYP3A4 at clinically relevant concentrations. Venetoclax is a weak inhibitor of CYP2C8, CYP2C9 and UGT1A1 in vitro, but it is not predicted to cause clinically relevant inhibition. Venetoclax is not an inhibitor of UGT1A4, UGT1A6, UGT1A9 and UGT2B7.
Co-administration with transporter substrates/inhibitors:
Venetoclax is a P-gp and BCRP substrate as well as a P-gp and BCRP inhibitor and a weak OATP1B1 inhibitor in vitro (see section 4.5). Venetoclax is not expected to inhibit OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, or MATE2K at clinically relevant concentrations.
Elimination
The population estimate for the terminal phase elimination half-life of venetoclax was approximately 26 hours. Venetoclax shows minimal accumulation with accumulation ratio of 1.30-1.44. After a single oral administration of 200 mg radiolabeled [14C]-venetoclax to healthy subjects, >99.9% of the dose was recovered in faeces and <0.1% of the dose was excreted in urine within 9 days. Unchanged venetoclax accounted for 20.8% of the administered radioactive dose excreted in faeces. The pharmacokinetics of venetoclax do not change over time.
Special populations
Renal impairment
Based on a population pharmacokinetic analysis that included 321 subjects with mild renal impairment (CrCl ≥60 and <90 ml/min), 219 subjects with moderate renal impairment (CrCl ≥30 and <60 ml/min), 5 subjects with severe renal impairment (CrCl ≥15 and <30 ml/min) and 224 subjects with normal renal function (CrCl ≥90 ml/min), venetoclax exposures in subjects with mild, moderate or severe renal impairment are similar to those with normal renal function. The pharmacokinetics of venetoclax has not been studied in subjects with CrCl <15 ml/min or patients on dialysis (see section 4.2).
Hepatic impairment
Based on a population pharmacokinetic analysis that included 74 subjects with mild hepatic impairment, 7 subjects with moderate hepatic impairment and 442 subjects with normal hepatic function, venetoclax exposures are similar in subjects with mild and moderate hepatic impairment and normal hepatic function. Mild hepatic impairment was defined as normal total bilirubin and aspartate transaminase (AST) > upper limit of normal (ULN) or total bilirubin >1.0 to 1.5 times ULN, moderate hepatic impairment as total bilirubin >1.5 to 3.0 times ULN, and severe hepatic impairment as total bilirubin >3.0 ULN.
In a dedicated hepatic impairment study, venetoclax Cmax and AUC in subjects with mild (Child-Pugh A; n=6) or moderate (Child-Pugh B; n=6) hepatic impairment were similar to subjects with normal hepatic function, after receiving a 50 mg single dose of venetoclax. In subjects with severe (Child-Pugh C; n=5) hepatic impairment, the mean venetoclax Cmax was similar to subjects with normal hepatic function but venetoclax AUCinf was on average 2.7-fold higher (range: no change to 5-fold higher) than venetoclax AUCinf in the subjects with normal hepatic function (see section 4.2).
Effects of age, sex, weight and race
Based on population pharmacokinetic analyses, age, sex, and weight do not have an effect on venetoclax clearance. The exposure is 67% higher in Asian subjects as compared to non-Asian subjects. This difference is not considered clinically relevant.
Preclinical safety data
Toxicities observed in animal studies with venetoclax included dose-dependent reductions in lymphocytes and red blood cell mass. Both effects were reversible after cessation of dosing with venetoclax, with recovery of lymphocytes occurring 18 weeks post treatment. Both B- and T-cells were affected, but the most significant decreases occurred with B-cells.
Venetoclax also caused single cell necrosis in various tissues, including the gallbladder and exocrine pancreas, with no evidence of disruption of tissue integrity or organ dysfunction; these findings were minimal to mild in magnitude.
After approximately 3 months of daily dosing in dogs, venetoclax caused progressive white discoloration of the hair coat, due to loss of melanin pigment in the hair.
Carcinogenicity/genotoxicity
Venetoclax and the M27 major human metabolite were not carcinogenic in a 6-month transgenic (Tg.rasH2) mouse carcinogenicity study at oral doses up to 400 mg/kg/day of venetoclax and at a single dose level of 250 mg/kg/day of M27. Exposure margins (AUC), relative to the clinical AUC at 400 mg/day, were approximately 2-fold for venetoclax and 5.8-fold for M27.
Venetoclax was not genotoxic in bacterial mutagenicity assay, in vitro chromosome aberration assay and in vivo mouse micronucleus assay. The M27 metabolite was negative for genotoxicity in the bacterial mutagenicity and chromosomal aberration assays.
Reproductive toxicity
No effects on fertility were observed in fertility and early embryonic development studies in male and female mice. Testicular toxicity (germ cell loss) was observed in general toxicity studies in dogs at exposures of 0.5 to 18 times the human AUC exposure at a dose of 400 mg. Reversibility of this finding has not been demonstrated.
In embryo-foetal development studies in mice, venetoclax was associated with increased post-implantation loss and decreased foetal body weight at exposures of 1.1 times the human AUC exposure at a dose of 400 mg. The major human metabolite M27 was associated with post- implantation loss and resorptions at exposures approximately 9-times the human M27-AUC exposure at a 400 mg dose of venetoclax. In rabbits, venetoclax produced maternal toxicity, but no foetal toxicity at exposures of 0.1 times the human AUC exposure at a 400 mg dose.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.