VFEND Powder for solution Ref.[8126] Active ingredients: Voriconazole
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium
Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Coadministration with CYP3A4 substrates, terfenadine, astemizole, cisapride, pimozide or quinidine since increased plasma concentrations of these medicinal products can lead to QTc prolongation and rare occurrences of torsades de pointes (see section 4.5).
Coadministration with rifampicin, carbamazepine and phenobarbital since these medicinal products are likely to decrease plasma voriconazole concentrations significantly (see section 4.5).
Coadministration of standard doses of voriconazole with efavirenz doses of 400 mg once daily or higher is contraindicated, because efavirenz significantly decreases plasma voriconazole concentrations in healthy subjects at these doses. Voriconazole also significantly increases efavirenz plasma concentrations (see section 4.5, for lower doses see section 4.4).
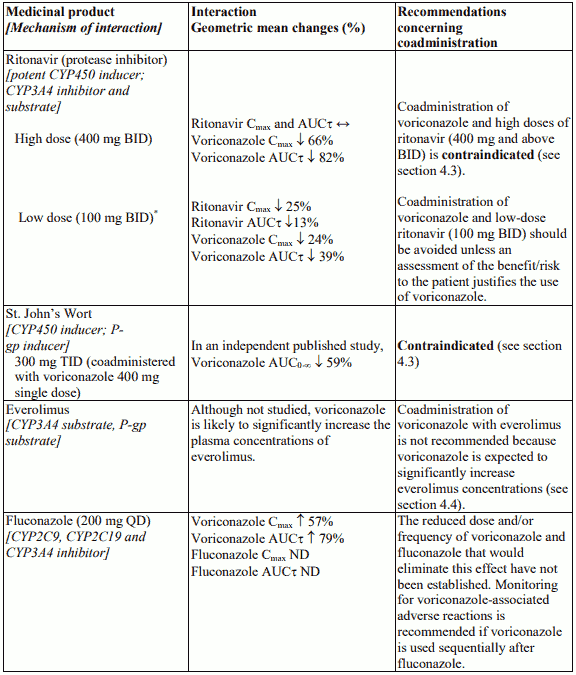
Coadministration with high-dose ritonavir (400 mg and above twice daily) because ritonavir significantly decreases plasma voriconazole concentrations in healthy subjects at this dose (see section 4.5, for lower doses see section 4.4).
Coadministration with ergot alkaloids (ergotamine, dihydroergotamine), which are CYP3A4 substrates, since increased plasma concentrations of these medicinal products can lead to ergotism (see section 4.5).
Coadministration with sirolimus since voriconazole is likely to increase plasma concentrations of sirolimus significantly (see section 4.5).
Coadministration with St. John’s Wort (see section 4.5).
Special warnings and precautions for use
Hypersensitivity
Caution should be used in prescribing VFEND to patients with hypersensitivity to other azoles (see also section 4.8).
Duration of treatment
The duration of treatment with the intravenous formulation should be no longer than 6 months (see section 5.3).
Cardiovascular
Voriconazole has been associated with QTc interval prolongation. There have been rare cases of torsades de pointes in patients taking voriconazole who had risk factors, such as history of cardiotoxic chemotherapy, cardiomyopathy, hypokalaemia and concomitant medicinal products that may have been contributory. Voriconazole should be administered with caution to patients with potentially proarrhythmic conditions, such as:
- Congenital or acquired QTc-prolongation.
- Cardiomyopathy, in particular when heart failure is present.
- Sinus bradycardia.
- Existing symptomatic arrhythmias.
- Concomitant medicinal product that is known to prolong QTc interval. Electrolyte disturbances such as hypokalaemia, hypomagnesaemia and hypocalcaemia should be monitored and corrected, if necessary, prior to initiation and during voriconazole therapy (see section 4.2). A study has been conducted in healthy volunteers which examined the effect on QTc interval of single doses of voriconazole up to 4 times the usual daily dose. No subject experienced an interval exceeding the potentially clinically-relevant threshold of 500 msec (see section 5.1).
Infusion-related reactions
Infusion-related reactions, predominantly flushing and nausea, have been observed during administration of the intravenous formulation of voriconazole. Depending on the severity of symptoms, consideration should be given to stopping treatment (see section 4.8).
Hepatic toxicity
In clinical trials, there have been cases of serious hepatic reactions during treatment with voriconazole (including clinical hepatitis, cholestasis and fulminant hepatic failure, including fatalities). Instances of hepatic reactions were noted to occur primarily in patients with serious underlying medical conditions (predominantly haematological malignancy). Transient hepatic reactions, including hepatitis and jaundice, have occurred among patients with no other identifiable risk factors. Liver dysfunction has usually been reversible on discontinuation of therapy (see section 4.8).
Monitoring of hepatic function
Patients receiving VFEND must be carefully monitored for hepatic toxicity. Clinical management should include laboratory evaluation of hepatic function (specifically AST and ALT) at the initiation of treatment with VFEND and at least weekly for the first month of treatment. Treatment duration should be as short as possible; however, if based on the benefit-risk assessment the treatment is continued (see section 4.2), monitoring frequency can be reduced to monthly if there are no changes in the liver function tests.
If the liver function tests become markedly elevated, VFEND should be discontinued, unless the medical judgment of the risk-benefit of the treatment for the patient justifies continued use.
Monitoring of hepatic function should be carried out in both children and adults.
Serious dermatological adverse reactions
Phototoxicity
In addition VFEND has been associated with phototoxicity including reactions such as ephelides, lentigo, actinic keratosis and pseudoporphyria. It is recommended that all patients, including children, avoid exposure to direct sunlight during VFEND treatment and use measures such as protective clothing and sunscreen with high sun protection factor (SPF).
Squamous cell carcinoma of the skin (SCC)
Squamous cell carcinoma of the skin has been reported in patients, some of whom have reported prior phototoxic reactions. If phototoxic reactions occur multidisciplinary advice should be sought, VFEND discontinuation and use of alternative antifungal agents should be considered and the patient should be referred to a dermatologist. If VFEND is continued, however, dermatologic evaluation should be performed on a systematic and regular basis, to allow early detection and management of premalignant lesions. VFEND should be discontinued if premalignant skin lesions or squamous cell carcinoma are identified (see below the section under Long-term treatment).
Exfoliative cutaneous reactions
Severe cutaneous adverse reactions (SCARs) such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS), which can be life-threatening or fatal, have been reported with the use of voriconazole. If a patient develops a rash he should be monitored closely and VFEND discontinued if lesions progress.
Long-term treatment
Long term exposure (treatment or prophylaxis) greater than 180 days (6 months) requires careful assessment of the benefit-risk balance and physicians should therefore consider the need to limit the exposure to VFEND (see sections 4.2 and 5.1).
Squamous cell carcinoma of the skin (SCC) has been reported in relation with long-term VFEND treatment.
Non-infectious periostitis with elevated fluoride and alkaline phosphatase levels has been reported in transplant patients. If a patient develops skeletal pain and radiologic findings compatible with periostitis VFEND discontinuation should be considered after multidisciplinary advice.
Visual adverse reactions
There have been reports of prolonged visual adverse reactions, including blurred vision, optic neuritis and papilloedema (see section 4.8).
Renal adverse reactions
Acute renal failure has been observed in severely ill patients undergoing treatment with VFEND. Patients being treated with voriconazole are likely to be treated concomitantly with nephrotoxic medicinal products and have concurrent conditions that may result in decreased renal function (see section 4.8).
Monitoring of renal function
Patients should be monitored for the development of abnormal renal function. This should include laboratory evaluation, particularly serum creatinine.
Monitoring of pancreatic function
Patients, especially children, with risk factors for acute pancreatitis (e.g., recent chemotherapy, haematopoietic stem cell transplantation [HSCT]), should be monitored closely during VFEND treatment. Monitoring of serum amylase or lipase may be considered in this clinical situation.
Paediatric population
Safety and effectiveness in paediatric subjects below the age of two years has not been established (see sections 4.8 and 5.1). Voriconazole is indicated for paediatric patients aged two years or older. A higher frequency of liver enzyme elevations was observed in the paediatric population (see section 4.8). Hepatic function should be monitored in both children and adults. Oral bioavailability may be limited in paediatric patients aged 2 to <12 years with malabsorption and very low body weight for age. In that case, intravenous voriconazole administration is recommended.
Serious dermatological adverse reactions (including SCC)
The frequency of phototoxicity reactions is higher in the paediatric population. As an evolution towards SCC has been reported, stringent measures for the photoprotection are warranted in this population of patients. In children experiencing photoaging injuries such as lentigines or ephelides, sun avoidance and dermatologic follow-up are recommended even after treatment discontinuation.
Prophylaxis
In case of treatment-related adverse events (hepatotoxicity, severe skin reactions including phototoxicity and SCC, severe or prolonged visual disorders and periostitis), discontinuation of voriconazole and use of alternative antifungal agents must be considered.
Phenytoin (CYP2C9 substrate and potent CYP450 inducer)
Careful monitoring of phenytoin levels is recommended when phenytoin is coadministered with voriconazole. Concomitant use of voriconazole and phenytoin should be avoided unless the benefit outweighs the risk (see section 4.5).
Efavirenz (CYP450 inducer; CYP3A4 inhibitor and substrate)
When voriconazole is coadministered with efavirenz the dose of voriconazole should be increased to 400 mg every 12 hours and the dose of efavirenz should be decreased to 300 mg every 24 hours (see sections 4.2, 4.3 and 4.5).
Rifabutin (Potent CYP450 inducer)
Careful monitoring of full blood counts and adverse reactions to rifabutin (e.g., uveitis) is recommended when rifabutin is coadministered with voriconazole. Concomitant use of voriconazole and rifabutin should be avoided unless the benefit outweighs the risk (see section 4.5).
Ritonavir (potent CYP450 inducer; CYP3A4 inhibitor and substrate)
Coadministration of voriconazole and low-dose ritonavir (100 mg twice daily) should be avoided unless an assessment of the benefit/risk to the patient justifies the use of voriconazole (see sections 4.3 and 4.5).
Everolimus (CYP3A4 substrate, P-gp substrate)
Coadministration of voriconazole with everolimus is not recommended because voriconazole is expected to significantly increase everolimus concentrations. Currently there are insufficient data to allow dosing recommendations in this situation (see section 4.5).
Methadone (CYP3A4 substrate)
Frequent monitoring for adverse reactions and toxicity related to methadone, including QTc prolongation, is recommended when coadministered with voriconazole since methadone levels increased following coadministration of voriconazole. Dose reduction of methadone may be needed (see section 4.5).
Short-acting opiates (CYP3A4 substrate)
Reduction in the dose of alfentanil, fentanyl and other short-acting opiates similar in structure to alfentanil and metabolised by CYP3A4 (e.g., sufentanil) should be considered when coadministered with voriconazole (see section 4.5). As the half-life of alfentanil is prolonged in a 4-fold manner when alfentanil is coadministered with voriconazole, and in an independent published study concomitant use of voriconazole with fentanyl resulted in an increase in the mean AUC0-∞ of fentanyl, frequent monitoring for opiateassociated adverse reactions (including a longer respiratory monitoring period) may be necessary.
Long-acting opiates (CYP3A4 substrate)
Reduction in the dose of oxycodone and other long-acting opiates metabolized by CYP3A4 (e.g., hydrocodone) should be considered when coadministered with voriconazole. Frequent monitoring for opiateassociated adverse reactions may be necessary (see section 4.5).
Fluconazole (CYP2C9, CYP2C19 and CYP3A4 inhibitor)
Coadministration of oral voriconazole and oral fluconazole resulted in a significant increase in Cmax and AUCτ of voriconazole in healthy subjects. The reduced dose and/or frequency of voriconazole and fluconazole that would eliminate this effect have not been established. Monitoring for voriconazoleassociated adverse reactions is recommended if voriconazole is used sequentially after fluconazole (see section 4.5).
Sodium content
Each vial of VFEND contains 217.6 mg of sodium. This should be taken into consideration for patients on a controlled sodium diet.
Interaction with other medicinal products and other forms of interaction
Voriconazole is metabolised by, and inhibits the activity of, cytochrome P450 isoenzymes, CYP2C19, CYP2C9, and CYP3A4. Inhibitors or inducers of these isoenzymes may increase or decrease voriconazole plasma concentrations, respectively, and there is potential for voriconazole to increase the plasma concentrations of substances metabolised by these CYP450 isoenzymes.
Unless otherwise specified, drug interaction studies have been performed in healthy adult male subjects using multiple dosing to steady state with oral voriconazole at 200 mg twice daily (BID). These results are relevant to other populations and routes of administration.
Voriconazole should be administered with caution in patients with concomitant medication that is known to prolong QTc interval. When there is also a potential for voriconazole to increase the plasma concentrations of substances metabolised by CYP3A4 isoenzymes (certain antihistamines, quinidine, cisapride, pimozide), coadministration is contraindicated (see below and section 4.3).
Interaction table
Interactions between voriconazole and other medicinal products are listed in the table below (once daily as “QD”, twice daily as “BID”, three times daily as “TID” and not determined as “ND”). The direction of the arrow for each pharmacokinetic parameter is based on the 90% confidence interval of the geometric mean ratio being within (↔), below (↓) or above (↑) the 80-125% range. The asterisk (*) indicates a twoway interaction. AUCτ, AUCt and AUC0-∞ represent area under the curve over a dosing interval, from time zero to the time with detectable measurement and from time zero to infinity, respectively.
The interactions in the table are presented in the following order: contraindications, those requiring dose adjustment and careful clinical and/or biological monitoring, and finally those that have no significant pharmacokinetic interaction but may be of clinical interest in this therapeutic field.





Fertility, pregnancy and lactation
Pregnancy
There are no adequate data on the use of VFEND in pregnant women available.
Studies in animals have shown reproductive toxicity (see section 5.3). The potential risk for humans is unknown.
VFEND must not be used during pregnancy unless the benefit to the mother clearly outweighs the potential risk to the foetus.
Women of child-bearing potential
Women of child-bearing potential must always use effective contraception during treatment.
Breast-feeding
The excretion of voriconazole into breast milk has not been investigated. Breast-feeding must be stopped on initiation of treatment with VFEND.
Fertility
In an animal study, no impairment of fertility was demonstrated in male and female rats (see section 5.3).
Effects on ability to drive and use machines
VFEND has moderate influence on the ability to drive and use machines. It may cause transient and reversible changes to vision, including blurring, altered/enhanced visual perception and/or photophobia. Patients must avoid potentially hazardous tasks, such as driving or operating machinery while experiencing these symptoms.
Undesirable effects
Summary of safety profile
The safety profile of voriconazole in adults is based on an integrated safety database of more than 2,000 subjects (including 1,603 adult patients in therapeutic trials) and an additional 270 adults in prophylaxis trials. This represents a heterogeneous population, containing patients with haematological malignancy, HIV-infected patients with oesophageal candidiasis and refractory fungal infections, non-neutropenic patients with candidaemia or aspergillosis and healthy volunteers.
The most commonly reported adverse reactions were visual impairment, pyrexia, rash, vomiting, nausea, diarrhoea, headache, peripheral oedema, liver function test abnormal, respiratory distress and abdominal pain.
The severity of the adverse reactions was generally mild to moderate. No clinically significant differences were seen when the safety data were analysed by age, race, or gender.
Tabulated list of adverse reactions
In the table below, since the majority of the studies were of an open nature, all causality adverse reactions and their frequency categories in 1,873 adults from pooled therapeutic (1,603) and prophylaxis (270) studies, by system organ class, are listed.
Frequency categories are expressed as: Very common (≥1/10); Common (≥1/100 to <1/10); Uncommon (≥1/1,000 to <1/100); Rare (≥1/10,000 to <1/1,000); Very rare (<1/10,000); Not known (cannot be estimated from the available data).
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Undesirable effects reported in subjects receiving voriconazole:
Infections and infestations
Common: sinusitis
Uncommon: pseudomembranous colitis
Neoplasms benign, malignant and unspecified (including cysts and polyps)
Frequency not known: squamous cell carcinoma*
Blood and lymphatic system disorders
Common: agranulocytosis1, pancytopenia, thrombocytopenia2, leukopenia, anaemia
Uncommon: bone marrow failure, lymphadenopathy, eosinophilia
Rare: disseminated intravascular coagulation
Immune system disorders
Uncommon: hypersensitivity
Rare: anaphylactoid reaction
Endocrine disorders
Uncommon: adrenal insufficiency, hypothyroidism
Rare: hyperthyroidism
Metabolism and nutrition disorders
Very common: oedema peripheral
Common: hypoglycaemia, hypokalaemia, hyponatraemia
Psychiatric disorders
Common: depression, hallucination, anxiety, insomnia, agitation, confusional state
Nervous system disorders
Very common: headache
Common: convulsion, syncope, tremor, hypertonia3, paraesthesia, somnolence, dizziness
Uncommon: brain oedema, encephalopathy4, extrapyramidal disorder5, neuropathy peripheral, ataxia, hypoaesthesia, dysgeusia
Rare: hepatic encephalopathy, Guillain-Barre syndrome, nystagmus
Eye disorders
Very common: visual impairment6
Common: retinal haemorrhage
Uncommon: optic nerve disorder7, papilloedema8, oculogyric crisis, diplopia, scleritis, blepharitis
Rare: optic atrophy, corneal opacity
Ear and labyrinth disorders
Uncommon: hypoacusis, vertigo, tinnitus
Cardiac disorders
Common: arrhythmia supraventricular, tachycardia, bradycardia
Uncommon: ventricular fibrillation, ventricular extrasystoles, ventricular tachycardia, electrocardiogram QT prolonged, supraventricular tachycardia
Rare: torsades de pointes, atrioventricular block complete, bundle branch block, nodal rhythm
Vascular disorders
Common: hypotension, phlebitis
Uncommon: thrombophlebitis, lymphangitis
Respiratory, thoracic and mediastinal disorders
Very common: respiratory distress9
Common: acute respiratory distress syndrome, pulmonary oedema
Gastrointestinal disorders
Very common: diarrhoea, vomiting, abdominal pain, nausea
Common: cheilitis, dyspepsia, constipation, gingivitis
Uncommon: peritonitis, pancreatitis, swollen tongue, duodenitis, gastroenteritis, glossitis
Hepatobiliary disorders
Very common: liver function test abnormal
Common: jaundice, jaundice cholestatic, hepatitis10
Uncommon: hepatic failure, hepatomegaly, cholecystitis, cholelithiasis
Skin and subcutaneous tissue disorders
Very common: rash
Common: dermatitis exfoliative, alopecia, rash maculo-papular, pruritus, erythema
Uncommon: Stevens-Johnson syndrome8, phototoxicity, purpura, urticaria, dermatitis allergic, rash papular, rash macular, eczema
Rare: toxic epidermal necrolysis8, drug reaction with eosinophilia and systemic symptoms (DRESS)8, angioedema, actinic keratosis*, pseudoporphyria, erythema multiforme, psoriasis, drug eruption
Frequency not known: cutaneous lupus erythematosus*, ephelides*, lentigo*
Musculoskeletal and connective tissue disorders
Common: back pain
Uncommon: arthritis
Frequency not known: periostitis*
Renal and urinary disorders
Common: renal failure acute, haematuria
Uncommon: renal tubular necrosis, proteinuria, nephritis
General disorders and administration site conditions
Very common: pyrexia
Common: chest pain, face oedema11, asthenia, chills
Uncommon: infusion site reaction, influenza like illness
Investigations
Common: blood creatinine increased
Uncommon: blood urea increased, blood cholesterol increased
* ADR identified post-marketing
1 Includes febrile neutropenia and neutropenia.
2 Includes immune thrombocytopenic purpura.
3 Includes nuchal rigidity and tetany.
4 Includes hypoxic-ischaemic encephalopathy and metabolic encephalopathy.
5 Includes akathisia and parkinsonism.
6 See “Visual impairments” paragraph in section 4.8.
7 Prolonged optic neuritis has been reported post-marketing. See section 4.4.
8 See section 4.4.
9 Includes dyspnoea and dyspnoea exertional.
10 Includes drug-induced liver injury, hepatitis toxic, hepatocellular injury and hepatotoxicity.
11 Includes periorbital oedema, lip oedema, and oedema mouth.
Description of selected adverse reactions
Visual impairments
In clinical trials, visual impairments (including blurred vision, photophobia, chloropsia, chromatopsia, colour blindness, cyanopsia, eye disorder, halo vision, night blindness, oscillopsia, photopsia, scintillating scotoma, visual acuity reduced, visual brightness, visual field defect, vitreous floaters, and xanthopsia) with voriconazole were very common. These visual impairments were transient and fully reversible, with the majority spontaneously resolving within 60 minutes and no clinically significant long-term visual effects were observed. There was evidence of attenuation with repeated doses of voriconazole. The visual impairments were generally mild, rarely resulted in discontinuation and were not associated with long-term sequelae. Visual impairments may be associated with higher plasma concentrations and/or doses.
The mechanism of action is unknown, although the site of action is most likely to be within the retina. In a study in healthy volunteers investigating the impact of voriconazole on retinal function, voriconazole caused a decrease in the electroretinogram (ERG) waveform amplitude. The ERG measures electrical currents in the retina. The ERG changes did not progress over 29 days of treatment and were fully reversible on withdrawal of voriconazole.
There have been post-marketing reports of prolonged visual adverse events (see section 4.4).
Dermatological reactions
Dermatological reactions were very common in patients treated with voriconazole in clinical trials, but these patients had serious underlying diseases and were receiving multiple concomitant medicinal products. The majority of rashes were of mild to moderate severity. Patients have developed severe cutaneous adverse reactions (SCARs), including Stevens-Johnson syndrome (SJS) (uncommon), toxic epidermal necrolysis (TEN) (rare), drug reaction with eosinophilia and systemic symptoms (DRESS) (rare) and erythema multiforme (rare) during treatment with VFEND (see section 4.4).
If a patient develops a rash they should be monitored closely and VFEND discontinued if lesions progress. Photosensitivity reactions such as ephelides, lentigo and actinic keratosis have been reported, especially during long-term therapy (see section 4.4).
There have been reports of squamous cell carcinoma of the skin in patients treated with VFEND for long periods of time; the mechanism has not been established (see section 4.4).
Liver function tests
The overall incidence of transaminase increases >3 xULN (not necessarily comprising an adverse event) in the voriconazole clinical programme was 18.0% (319/1,768) in adults and 25.8% (73/283) in paediatric subjects who received voriconazole for pooled therapeutic and prophylaxis use. Liver function test abnormalities may be associated with higher plasma concentrations and/or doses. The majority of abnormal liver function tests either resolved during treatment without dose adjustment or following dose adjustment, including discontinuation of therapy.
Voriconazole has been associated with cases of serious hepatic toxicity in patients with other serious underlying conditions. This includes cases of jaundice, hepatitis and hepatic failure leading to death (see section 4.4).
Infusion-related reactions
During infusion of the intravenous formulation of voriconazole in healthy subjects, anaphylactoid-type reactions, including flushing, fever, sweating, tachycardia, chest tightness, dyspnoea, faintness, nausea, pruritus and rash have occurred. Symptoms appeared immediately upon initiating the infusion (see section 4.4).
Prophylaxis
In an open-label, comparative, multicenter study comparing voriconazole and itraconazole as primary prophylaxis in adult and adolescent allogeneic HSCT recipients without prior proven or probable IFI, permanent discontinuation of voriconazole due to AEs was reported in 39.3% of subjects versus 39.6% of subjects in the itraconazole arm. Treatment-emergent hepatic AEs resulted in permanent discontinuation of study medication for 50 subjects (21.4%) treated with voriconazole and for 18 subjects (7.1%) treated with itraconazole.
Paediatric population
The safety of voriconazole was investigated in 288 paediatric patients aged 2 to <12 years (169) and 12 to <18 years (119) who received voriconazole for prophylaxis (183) and therapeutic use (105) in clinical trials. The safety of voriconazole was also investigated in 158 additional paediatric patients aged 2 to <12 years in compassionate use programs. Overall, the safety profile of voriconazole in paediatric population was similar to that in adults. However, a trend towards a higher frequency of liver enzyme elevations, reported as adverse events in clinical trials was observed in paediatric patients as compared to adults (14.2% transaminases increased in paediatrics compared to 5.3% in adults). Post-marketing data suggest there might be a higher occurrence of skin reactions (especially erythema) in the paediatric population compared to adults. In the 22 patients less than 2 years old who received voriconazole in a compassionate use programme, the following adverse reactions (for which a relationship to voriconazole could not be excluded) were reported: photosensitivity reaction (1), arrhythmia (1), pancreatitis (1), blood bilirubin increased (1), hepatic enzymes increased (1), rash (1) and papilloedema (1). There have been post-marketing reports of pancreatitis in paediatric patients.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
VFEND must not be infused into the same line or cannula concomitantly with other intravenous products. The bag should be checked to ensure that the infusion is complete. When the VFEND infusion is complete, the line may be used for administration of other intravenous products.
Blood products and short-term infusion of concentrated solutions of electrolytes
Electrolyte disturbances such as hypokalaemia, hypomagnesaemia and hypocalcaemia should be corrected prior to initiation of voriconazole therapy (see sections 4.2 and 4.4). VFEND must not be administered simultaneously with any blood product or any short-term infusion of concentrated solutions of electrolytes, even if the two infusions are running in separate lines.
Total parenteral nutrition
Total parenteral nutrition (TPN) need not be discontinued when prescribed with VFEND, but does need to be infused through a separate line. If infused through a multiple-lumen catheter, TPN needs to be administered using a different port from the one used for VFEND. VFEND must not be diluted with 4.2% Sodium Bicarbonate Infusion. Compatibility with other concentrations is unknown.
This medicinal product must not be mixed with other medicinal products except those mentioned in section 6.6.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.