WELIREG Film-coated tablet Ref.[114790] Active ingredients: Belzutifan
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Merck Sharp & Dohme B.V., Waarderweg 39, 2031 BN Haarlem, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agent, other antineoplastic agents
ATC code: L01XX74
Mechanism of action
Belzutifan is an inhibitor of the transcription factor hypoxia-inducible factor 2 alpha (HIF-2α). Under normal oxygen levels, HIF-2α is targeted for degradation by VHL protein. Impairment of VHL protein function results in accumulation of HIF-2α. Consequently, HIF-2α translocates into the nucleus and regulates expression genes, associated with cellular proliferation, angiogenesis, and tumour growth. Belzutifan binds to HIF-2α, and in conditions of hypoxia or impairment of VHL protein function, belzutifan blocks the HIF-2α-HIF-1β interaction, leading to reduced transcription and expression of HIF-2α target genes.
Pharmacodynamic effects
Circulating plasma levels of erythropoietin (EPO) were monitored in patients as a pharmacodynamic marker of HIF-2α inhibition. Reductions in EPO were observed to be dose/exposure dependent and showed a plateauing effect on reduction at exposures achieved with doses above 120 mg once daily. The maximum EPO suppression occurred following 2 weeks of consecutive dosing of belzutifan (mean percent decrease from baseline of approximately 60%). Mean EPO levels gradually returned to baseline values after 12 weeks of treatment.
At the recommended dose (120 mg once daily) for belzutifan, there were no clinically relevant effects on the QTc interval.
Clinical efficacy
Clinical study in adult patients with advanced renal cell carcinoma (RCC)
The efficacy of belzutifan was evaluated in LITESPARK-005, an open-label, randomised, active-controlled Phase 3 clinical study comparing belzutifan with everolimus in 746 patients with unresectable, locally advanced or metastatic clear cell RCC that has progressed following PD-1/L1 checkpoint inhibitors and VEGF receptor targeted therapies either in sequence or in combination.
Patients could have received up to 3 prior treatment regimens and must have measurable disease per RECIST v1.1. The study excluded patients with hypoxia, active CNS metastases and clinically significant cardiac disease. Patients were randomised in a 1:1 ratio to receive 120 mg belzutifan or 10 mg everolimus by oral administration once daily. Randomisation was stratified by International Metastatic RCC Database Consortium (IMDC) risk categories (favo urable versus intermediate versus poor) and number of prior VEGF receptor targete d therapies (1 versus 2-3).
Patients were evaluated radiologically at Week 9 from the date of randomi sation, then every 8 weeks through Week 49, and every 12 weeks thereafter.
Among the 746 patients in LITESPARK-005, 369 patients received two or more lines of therapy that included a PD-(L)1 inhibitor and at least two VEGF -targeted therapies. The baseline characteristics of those patients were: median age 63 years (range: 33 to 82 years), 40% age 65 or older; 11% age 75 or older; 79% male; 78% White; 12% Asian; 1% Black or African American; 42% ECOG performance status 0 and 56% ECOG performance status 1. Prior lines of therapies: 17% of patients had 2, 81% had 3 and 2% had 4 prior lines of therapies. Patient distribution by IMDC risk categories was 22% favourable, 66% intermediate, and 12% poor.
The primary efficacy outcome measures were progression-free survival (PFS) measured by BICR using RECIST v1.1, and overall survival (OS). Secondary efficacy outcome measures included objective response rate (ORR) and duration of response (DOR) by BICR using RECIST v1.1. In the overall population, the study demonstrated statistically significant improvements of PFS (HR: 0.75 [95% CI 0.63, 0.90], p-Value 0.00077) and ORR (21.9% versus 3.5%, p-Value <0.00001) for patients randomised to belzutifan compared with everolimus at a pre-specified interim analysis (median follow-up time of 13.5 months [range: 0.2 to 31.8 months]).
Table 3 summarises key efficacy measures in the subgroup of patients that received two or more lines of therapy that included a PD-(L)1 inhibitor and at least two VEGF-targeted therapies in LITESPARK-005. The KM curves for PFS and OS are shown in Figures 1 and 2.
Table 3. Efficacy results (BICR assessment) in LITESPARK-005 for patients that received two or more lines of therapy that included a PD-(L)1 inhibitor and at least two VEGF-targeted therapies:
| Endpoint | Belzutifan n=187 | Everolimus n=182 |
|---|---|---|
| PFS* | ||
| Number of events, n (%) | 127 (67.9%) | 130 (71.4%) |
| Median† PFS in months (95% CI) | 4.6 (3.5, 7.3) | 5.4 (3.8, 6.5) |
| Hazard ratio‡ (95% CI) | 0.73 (0.57, 0.94) | |
| OS¶ | ||
| Number of events, n (%) | 128 (68.1%) | 125 (68.7%) |
| Median† OS in months (95% CI) | 21.8 (17.4, 25.8) | 18.1 (14.2, 23.9) |
| Hazard ratio‡ (95% CI) | 0.94 (0.74, 1.21) | |
| ORR* % (95% CI) | 24.1% (18.1, 30.8) | 3.3% (1.2, 7.0) |
| Complete response, n (%) | 5 (2.7%) | 0 (0%) |
| Partial response, n (%) | 40 (21.4%) | 6 (3.3%) |
| Response duration* | ||
| Median in months (range) | NR (1.9+, 23.1+) | 17.2 (3.8, 17.2) |
* Based on first pre-specified interim analysis (median follow-up time of 13.5 months)
† From product-limit (Kaplan- Meier) method for censored data
‡ Based on Cox regression model
¶ Based on final analysis (median follow-up time of 19.6 months)
+ Denotes ongoing response
NR = Not reached
The median time to response (TTR) was 3.7 months (range: 1.7-16.6) in the belzutifan arm and 3.0 months (range: 1.8-5.4) in the everolimus arm (median follow-up time of 13.5 months), in the subgroup of patients that received two or more lines of therapy that included a PD-L(1) inhibitor and at least two VEGF-targeted therapies in LITESPARK-005.
Figure 1. Kaplan-Meier curve for progression-free survival by treatment arm in LITESPARK-005 for patients that received two or more lines of therapy that included a PD-(L)1 inhibitor and at least two VEGF-targeted therapies*:
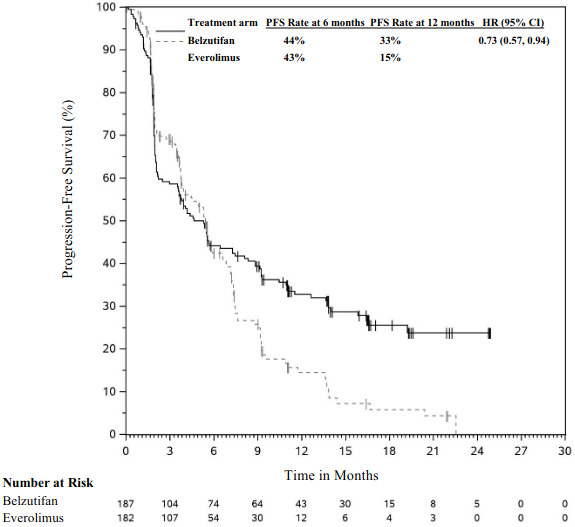
* Median follow-up time of 13.5 months
Figure 2. Kaplan-Meier curve for overall survival by treatment arm in LITESPARK-005 for patients that received two or more lines of therapy that included a PD-(L)1 inhibitor and at least two VEGF-targeted therapies*:
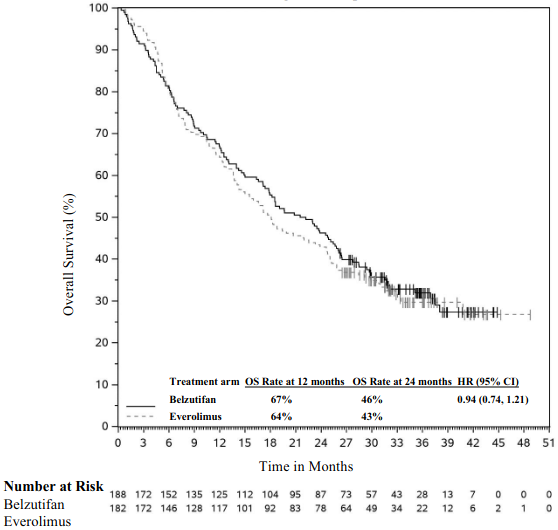
* Median follow-up time of 19.6 months
Clinical study in adult patients with von Hippel-Lindau (VHL) disease-associated tumours
The efficacy of belzutifan was investigated in LITESPARK-004, an open-label Phase 2 clinical study in 61 patients with VHL disease who had at least one measurable soli d tumour (as defined by RECIST v1.1) localised to the kidney and who did not require immediate surgery. Patients could also have other VHL disease-associated tumours, such as CNS haemangioblastomas and pNET. Patients received belzutifan at a dose of 120 mg once daily. Patients were evaluated radiologically approximately 12 weeks after initiation of treatment and every 12 weeks thereafter. Treatment was continued until progression of disease or unacceptable toxicity. Patients were required to have an ECOG PS of 0 or 1. The study excluded patients who had any evidence of metastatic disease, either RCC or other VHL disease-associated tumours, an immediate need for surgical intervention for tumour treatment, any major surgical procedure completed within 4 weeks prior to study enrolment, any major cardiovascular event within 6 months prior to study drug administration, or prior systemic treatments for VHL disease-associated RCC.
Among the 61 patients enrolled in LITESPARK-004, the population characteristics were: median age of 41 years, 3.3% age 65 or older; 52.5% male; 90.2% White; and 82.0% had an ECOG PS of 0 and 16.4% had an ECOG PS of 1. Seventy-seven percent of patients had prior RCC surgical procedures. Other VHL disease-associated tumours in patients included pancreatic lesions (100.0%) of which 36.1% were pancreatic neuroendocrine tumours, CNS haemangioblastomas (82.0%), and retinal angiomas (19.7%).
The primary efficacy endpoint for the treatment of VHL disease-associated RCC was ORR me asured by radiology assessment using RECIST v1.1 as assessed by a central independent review committee (IRC). Additional efficacy endpoints included DOR and TTR. ORR and DOR in other VHL disease-associated tumours were assessed as secondary efficacy endpoi nts.
Table 4 summarises the efficacy results for VHL disease -associated RCC tumours in LITESPARK-004, based on an interim analysis with a median follow-up time of 49.7 months.
Table 4. Efficacy results in VHL disease-associated RCC tumours in LITESPARK-004:
| Endpoint | Belzutifan n=61 |
|---|---|
| ORR* % (95% CI) | 67.2% (54.0, 78.7) |
| Complete response | 11.5% |
| Partial response | 55.7% |
| Response duration† | |
| Median in months (range) | NR (8.6+, 44.4+) |
| % with duration ≥12 months | 100.0% |
| Time to response | |
| Median in months (range) | 11.1 (2.7, 41.2) |
Efficacy data with a median follow -up of 49.7 months (cut-off date 3 Apr 2023)
* Response: Best objective response as confirmed complete response or partial response
† Based on Kaplan-Meier estimates
+ Denotes ongoing response
NR = Not reached
Efficacy endpoints for the treatment of other VHL disease -associated tumours included ORR and DOR, as assessed by IRC using RECIST v1.1. These results are sho wn in Table 5.
Table 5. Efficacy results for belzutifan for other VHL disease-associated tumours:
| Belzutifan n=61 | ||
|---|---|---|
| Endpoint | Patients with evaluable CNS haemangioblastomas n=50 | Patients with evaluable pancreatic neuroendocrine tumours n=22 |
| ORR* % (95% CI) | 48% (33.7, 62.6) | 90.9% (70.8, 98.9) |
| Complete response | 8.0% | 50.0% |
| Partial response | 40.0% | 40.9% |
| Response duration† | ||
| Median in months (range) | NR (0.0+, 47.5+) | NR (11.0+, 48.3+) |
| % with duration ≥12 months | 95.5% | 100.0% |
Efficacy data with a median follow-up of 49.7 months (cut-off date 3 Apr 2023)
* Response: Best objective response as confirmed complete response or partial response
† Based on Kaplan-Meier estimates
+ Denotes ongoing response
NR = Not reached
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with belzutifan in all subsets of the paediatric population in renal neoplasms and VHL disease (see section 4.2 for information on paediatric use).
Conditional approval
This medicinal product has been authorised under a so-called 'conditional approval' scheme. This means that further evidence on this medicinal product is awaited. The European Medicines Agency will review new information on this medicinal product at least every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
The pharmacokinetics of belzutifan are similar in healthy subjects and patients with solid tumours including advanced RCC. Based on population PK analysis, the simulated geometric mean steady-state (CV%) Cmax is 1.5 mcg/mL (46%) and AUC0-24hr is 20.8 mcg•hr/mL (64%) in patients treated with 120 mg belzutifan. Steady state is reached after approximately 3 days.
Absorption
Following single-dose oral administration of 120 mg of belzutifan, peak plasma concentrations (median Tmax) of belzutifan occurred at 1 to 2 hours post dose.
Effect of food
A high-fat, high-calorie meal delayed peak belzutifan concentration by approximately 2 hours but, had no effect on exposure (AUC). There was a modest decrease of Cmax by 24% following consumption of a high-fat, high-calorie meal, but this was not clinically meaningful. Therefore, belzutifan can be taken without regard to food.
Distribution
Based on the population PK analysis, the mean (CV%) volume of distribution is 120 L (28.5%). Plasma protein binding of belzutifan is 45%. The blood-to-plasma concentration ratio of belzutifan is 0.88.
Biotransformation
Belzutifan is primarily metabolised by UGT2B17 and CYP2C19 and to lesser extent by CYP3A4. Both UGT2B17 and CYP2C19 display genetic polymorphisms (see 'Special populations - Dual UGT2B17 and CYP2C19 Poor Metabolisers').
In vitro assessment of drug interactions
Belzutifan is a substrate of UGT2B17, CYP2C19 and CYP3A4. Active transport is not an important determinant of belzutifan disposition. Belzutifan is not an inhibitor of CYP enzymes, UGT enzymes, or transporters with the exception of MATE-2K, and potentially MATE1. Belzutifan does not induce CYP1A2, however, belzutifan induces CYP2B6, CYP2C8 and CYP3A4 in a concentration dependent manner (see section 4.5).
Elimination
Based on the population PK analysis, the mean (CV%) clearance is 5.89 L/hr (60.6%) and the mean elimination half-life is approximately 14 hrs.
Following oral administration of radiolabelled belzutifan to healthy subjects, approximately 49.6% of the dose was excreted in urine and 51.7% in faeces (primarily as inactive metabolites). Approximately 6% of the dose was recovered as parent drug in urine.
Linearity
The plasma Cmax and AUC increased proportionally over a dose range of 40 mg to 120 mg.
Special populations
Renal impairment
Based on a population pharmacokinetic analysis of belzutifan in healthy subjects and patients with cancer, no clinically significant differences in the mean belzutifan exposure were observed between subjects with normal renal function and those with mild and moderate renal impairment (as evaluated by estimated glomerular filtration rate (eGFR)). In a dedicated pharmacokinetic study, belzutifan exposure (AUC0-INF) decreased by 6% and increased by 14% in patients with end-stage renal disease before and after haemodialysis, respectively (see section 4.2).
Hepatic impairment
Based on a population pharmacokinetic analysis of belzutifan in healthy subjects and patients with cancer, no clinically significant differences in the mean belzutifan exposure were observed between subjects with normal liver function (total bilirubin and AST ≤ ULN), and those with mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin > 1 to 1.5 x ULN and any AST). In a dedicated pharmacokinetic study, belzutifan exposure (AUC0-INF) increased by 52% in patients with moderate hepatic impairment (Child-Pugh B). Patients with severe hepatic impairment have not been studied (see section 4.2).
Dual UGT2B17 and CYP2C19 Poor Metabolisers
Patients who are dual UGT2B17 and CYP2C19 poor metabolisers have higher belzutifan exposures, which may increase the incidence and severity of adverse reactions of belzutifan and should be closely monitored (see sections 4.4 and 4.8).
Belzutifan is primarily metabolised by UGT2B17 and CYP2C19. The activity of these enzymes varies among individuals who carry different genetic variants, which may impact belzutifan concentrations. Poor metabolisers are individuals who are considered to have no enzyme activity. In patients who are dual UGT2B17 and CYP2C19 poor metabolisers, CYP3A4 may be a major elimination pathway. Approximately 15% of Caucasians, 11% of Latinos, 6% of African Americans, 38% of South Asians, and 70% of East Asians are UGT2B17 poor metabolisers. Approximately 2% of Caucasians, 1% of Latinos, 5% of African Americans, 8% of South Asians, and 13% of East Asians are CYP2C19 poor metabolisers. Approximately 0.3% of Caucasians, 0.1% of Latinos, 0.3% of African Americans, 3% of South Asians, and 9% of East Asians are dual UGT2B17 and CYP2C19 poor metabolisers. Expected frequencies in the Japanese population for the UGT2B17, CYP2C19, and dual UGT2B17 and CYP2C19 poor metabolisers are approximately 77%, 19%, and 15%, respectively. Expected frequencies in the United States population for the UGT2B17, CYP2C19, and dual UGT2B17 and CYP2C19 poor metabolisers are approximately 16%, 3%, and 0.5%, respectively based on the reported proportion of the US population represented by major racial/ethnic groups.
The impact of CYP2C19 and UGT2B17 poor metabolisers on belzutifan exposure was assessed in a population PK analysis. Based on the population PK model, patients who are CYP2C19, UGT2B17, or dual UGT2B17 and CYP2C19 poor metabolisers, are projected to have 1.3-, 2.7- or 3.3-fold the exposures (steady-state AUC0-24hr), respectively, compared to a typical reference patient (UGT2B17 extensive metaboliser, CYP2C19 extensive/intermediate metaboliser) for the recommended dose. No dose adjustment is recommended based on exposure -response analyses for efficacy and safety and the risk-benefit profile.
Effects of age, gender, ethnicity, race, and body weight
Based on a population pharmacokinetic analysis, age (range: 19 to 90 years), gender, ethnicity, race, and body weight (range: 42.1 to 166 kg) do not have a clinically meaningful effect on the 16 pharmacokinetics of belzutifan. Potential differences in exposure across races are possible due to different frequencies of metaboli sing enzymes (see 'Special populations - Dual UGT2B17 and CYP2C19 Poor Metabolisers').
5.3. Preclinical safety data
Repeat dose toxicity
Repeat-dose oral toxicity studies in rats and dogs for up to 3 months duration revealed anaemia at all doses including at exposure levels lower than the human exposure levels. Although the anaemia was reversible, this is relevant to humans.
Carcinogenesis
A 26-week transgenic rasH2 mouse carcinogenicity study has been conducted with belzutifan at doses up to 600 mg/kg/day, corresponding to exposures up to 28-fold the human exposure at the approved dose. No belzutifan-related neoplastic findi ngs were observed at any dose level and no carcinogenic risk was identified in the study.
Mutagenesis
Belzutifan was not genotoxic in in vitro bacterial mutagenesis and micronucleus assays, and an in vivo rat micronucleus assay at 1.7-fold human exposure.
Reproductive toxicity
Fertility studies with belzutifan have not been conducted. In the 3-month repeat-dose toxicity study in rats, irreversible testicular atrophy/degeneration and oligospermia was observed at exposures lower than the human exp osure at the recommended dose of 120 mg daily. No testicular toxicity was observed in dogs up to an exposure similar to the human exposure. There were no findings in female reproductive organs in either rat or dog 3-month toxicity studies, but HIF-2α has a functional role in the uterus during embryo implantation and establishment of pregnancy in mice. HIF-2α inhibition by exposure to belzutifan has the potential to interfere with embryo implantation, leading to impairment of female fertility.
In a rat embryo-foetal development study, administration of belzutifan during organogenesis caused embryo-foetal lethality up to 100%, reduced foetal body weight, and foetal skeletal abnormalities at exposures similar to or below the human exposure at the recommended do se of 120 mg daily.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.