YSELTY Film-coated tablet Ref.[50145] Active ingredients: Linzagolix
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Theramex Ireland Limited, 3rd Floor, Kilmore House, Park Lane, Spencer Dock, Dublin 1, D01 YE64, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Anti-gonadotropin-releasing hormones
ATC code: H01CC04
Mechanism of action
Linzagolix is a selective, non-peptide gonadotropin-releasing hormone (GnRH) receptor antagonist that inhibits endogenous GnRH signalling by binding competitively to GnRH receptors in the pituitary gland, thereby modulating the hypothalamic-pituitary-gonadal axis.
Pharmacodynamic effects
Effects on pituitary and ovarian hormones
Administration of linzagolix results in dose-dependent suppression of luteinizing hormone and follicle-stimulating hormone, leading to decreased blood concentrations of estradiol and progesterone.
Uterine fibroid-treated population
In the phase 3 studies, full suppression of serum estradiol (median <20 pg/mL) was observed with linzagolix 200 mg from 4 to 24 weeks. Partial suppression was observed with linzagolix 100 mg, 100 mg with concomitant ABT (referred to as "with ABT") and 200 mg with ABT from 4 to 52 weeks, with median serum estradiol levels in the range of 20 to 60 pg/mL. Progesterone levels were maintained ≤3.1 ng/mL in 83% of women receiving linzagolix 200 mg for 24 weeks and 68% of women receiving linzagolix 100 mg for 52 weeks, and about 90% of women receiving linzagolix 100 mg with ABT or 200 mg with ABT for 52 weeks.
Endometriosis treated population
Median serum estradiol levels for subjects receiving 200 mg with concomitant ABT were in the range of 20 to 60 pg/mL.
Cardiac electrophysiology
One randomised, placebo- and positive-controlled, open-label, single-dose, crossover thorough-QTc study evaluated the effect of linzagolix on the QTc interval. Forty-eight healthy women received a 200 mg dose of linzagolix (therapeutic target exposure), a 700 mg dose of linzagolix (supratherapeutic target exposure), a 400 mg dose of moxifloxacin (positive control), or placebo with an appropriate washout. A marginal effect with linzagolix 200 mg and 700 mg doses on the prolongation of the heart-rate corrected QT interval was identified, with a maximum observed mean at 3 hours post dose of 8.34 msec (90% CI 6.44 - 10.23) and 9.92 msec (90% CI 8.03 - 11.81), respectively. Based on the magnitude of the QTc prolongation, subsequent concentration effect modelling and QT subinterval (JTpeakc), the observed effects are not considered clinically relevant. The highest anticipated steady state concentration in the QT study was estimated in healthy subjects, not accounting for increases in unbound linzagolix exposure due to existing disorders (see section 5.2).
Changes in lipid parameters
Fasting lipid levels (HDL, LDL and total cholesterol, and triglycerides) were assessed every three months from start of linzagolix treatment up to 3 months post treatment. There were increases in LDL cholesterol, HDL cholesterol, and triglycerides across all linzagolix arms (typically less than 15% in the case of LDL, and less than 20% in the case of triglycerides) and generally increases were higher for the linzagolix only regimes. These increases were evident from week 12 and lipid parameters had generally stabilised after 52 weeks of treatment. After stopping linzagolix, lipid levels showed signs of returning towards baseline by 12 weeks after stopping treatment, but still remained slightly elevated relative to baseline (see section 4.4).
Clinical efficacy and safety
Uterine fibroid-treated population
The efficacy of Yselty was evaluated in two phase 3, randomised, double-blind and placebo-controlled studies, PRIMROSE 1 and PRIMROSE 2, including 511 and 501 women, respectively. PRIMROSE 1 was conducted in the US and PRIMROSE 2 was conducted primarily in Europe with about 10% of subjects being from the US. The studies had essentially replicate design with 52 weeks of treatment and 24 weeks post treatment follow-up. There are no on-treatment efficacy or safety data beyond 52 weeks.
Eligible patients had heavy menstrual bleeding (HMB: >80 mL menstrual blood loss [MBL]/cycle) and a myomatous uterus with at least one fibroid ≥2 cm confirmed by ultrasound and no myoma >12 cm. MBL was measured using the alkaline haematin method.
The mean age of women was 42 years (range 20 to 58), and mean body mass index was 29.9 kg/m² (range 16.8 to 58.6). Approximately 34.5% of women were Black, 63.5% were White and 2% were of other races. The most commonly reported symptoms, in addition to HMB, were abdominal pain (67.9% of women), abdominal pressure (52.5%), menstruation lasting longer than usual (50.4%), lower back pain (50.2%), increased urinary frequency (34.5%) and pain during intercourse (27.7%). The median uterine volume was 241 cm³ (range 32 to 2075 cm³) and the median fibroid volume was 53 cm³ (range 0 to 1142 cm³). Almost all women (99.7%) had at least one fibroid ≥2 cm long and 97.5% had FIGO classification from 1 to 6.
Subjects were randomised to one of 5 treatments: placebo, Yselty 100 mg, Yselty 200 mg, Yselty 100 mg with concomitant ABT (estradiol 1 mg/ norethisterone acetate 0.5 mg, referred to as "with ABT") or Yselty 200 mg with ABT, all taken once daily. Subjects randomised to placebo or Yselty 200 mg were switched to Yselty 200 mg with ABT after 24 weeks except in PRIMROSE 1, in which 50% of placebo subjects continued placebo until 52 weeks.
The primary efficacy endpoint was a response, defined as having an MBL of ≤80 mL and ≥50% reduction from baseline over the last 28 days before week 24. Treatment with Yselty with or without ABT resulted in a higher proportion of women with reduced MBL at week 24 compared to placebo. The percentage of responders was 56.4%, 66.4%, 71.4% and 75.5% with Yselty 100 mg, 100 mg with ABT, 200 mg and 200 mg with ABT, respectively in PRIMROSE 1 and 56.7%, 77.2%, 77.7% and 93.9% respectively in PRIMROSE 2 (Table 3). At week 52, the percentage of responders was 57.4%, 79.9% and 87.9% with Yselty 100 mg, 100 mg with ABT and 200 mg with ABT, respectively, in PRIMROSE 1 and 53.2%, 91.3% and 91.6%, respectively, in PRIMROSE 2.
Table 3. Responders (women with reduced menstrual blood loss) at 24 weeks:
| Study | PRIMROSE 1 | PRIMROSE 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | Placebo | Yselty | Placebo | Yselty | ||||||
| 100 mg | 100 mg + ABT | 200 mg | 200 mg + ABT | 100 mg | 100 mg + ABT | 200 mg | 200 mg + ABT | |||
| N | 103 | 94 | 107 | 105 | 102 | 102 | 97 | 101 | 103 | 98 |
| Percentage (95% CI) of responders1,2 | 35.0 (25.8, 45.0) | 56.4 (45.8, 66.6) | 66.4 (56.6, 75.2) | 71.4 (61.8, 79.8) | 75.5 (66.0, 83.5) | 29.4 (20.8, 39.3) | 56.7 (46.3, 66.7) | 77.2 (67.8, 85.0) | 77.7 (68.4, 85.3) | 93.9 (87.1, 97.7) |
1 Responders were women with ≤ 80 mL MBL and ≥50% reduction from baseline
2 Clopper-Pearson 95% CI. p-values ≤0.003 for odds-ratio to placebo from a Cochran-Mantel-Haenszel test with race as stratification factor.
ABT: estradiol 1 mg/norethisterone acetate 0.5 mg
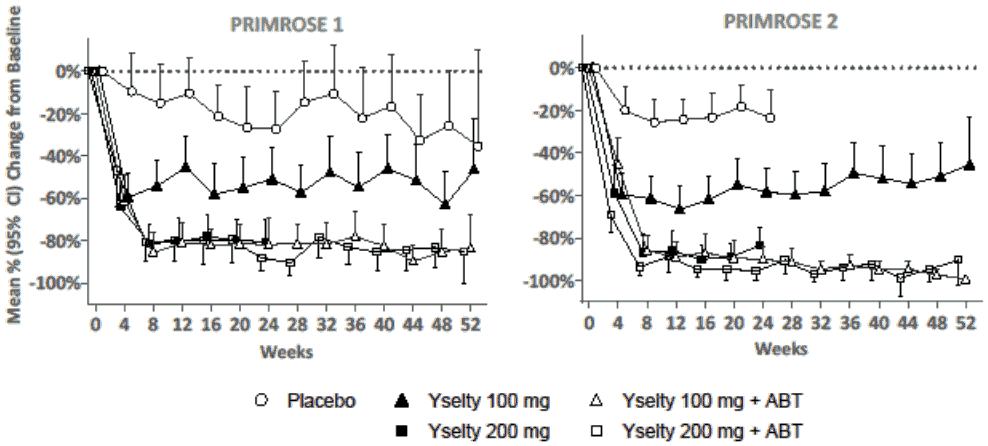
The mean percentage reduction in MBL over time is shown in Figure 1. Treatment with Yselty 100 mg achieved a maximal effect of about 60% reduction in MBL by 4 weeks. Treatment with Yselty 100 mg with ABT or 200 mg with or without ABT, reached a maximal effect of about 80 to 95% reduction in MBL by 8 weeks. These reductions were maintained up to 52 weeks.
Figure 1. Mean percentage change in menstrual blood loss for each 28-day period up to week 52:
In both pivotal phase 3 studies, improvements were observed in secondary endpoints after 24 weeks in the Yselty dose groups compared to placebo (Table 4), including an increased proportion of women achieving amenorrhea, reduced pain scores, higher haemoglobin levels in anaemic patients (<12 g/dL at baseline) and increased health-related quality of life scores. These improvements were more pronounced with Yselty 200 mg (with or without ABT) and Yselty 100 mg with ABT as compared to Yselty 100 mg.
Improvements in secondary endpoints at 24 weeks were generally maintained after 52 weeks in the Yselty 100 mg with and without ABT and Yselty 200 mg with ABT groups. Uterine and fibroid volumes were markedly and consistently reduced after 24 weeks, only in the Yselty 200 mg without ABT group. In PRIMROSE 1 and 2, respectively, uterine volumes were reduced by 31% and 43%, and fibroid volumes were reduced by 43% and 49%. Mean uterine and fibroid volumes increased toward baseline volumes when ABT was added after 6 months of treatment with Yselty 200 mg without ABT.
Table 4. Secondary endpoints at 24 weeks:
| Study | PRIMROSE 1 | PRIMROSE 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | Placebo | Yselty | Placebo | Yselty | ||||||
| 100 mg | 100 mg + ABT | 200 mg | 200 mg + ABT | 100 mg | 100 mg + ABT | 200 mg | 200 mg + ABT | |||
| N | 103 | 94 | 107 | 105 | 102 | 102 | 97 | 101 | 103 | 98 |
| Percentage of women with amenorrhea (95% CI)1 | 21.4 (13.9, 30.5) | 38.3 (28.5, 48.9) | 42.1 (32.6, 52.0) | 60.0 (50.0, 69.4) | 57.8 (47.7, 67.6) | 11.8 (6.2, 19.6) | 34.0 (24.7, 44.3) | 63.4 (53.2, 72.7) | 70.9 (61.1, 79.4) | 80.6 (71.4, 87.9) |
| Mean change from baseline in haemoglobin levels – g/dL (SD, n)2 | 0.30 (1.57, 45) | 1.36 (1.82, 42) | 1.87 (1.57, 52) | 2.22 (1.58, 53) | 2.00 (1.60, 50) | 0.38 (1.69, 43) | 1.36 (1.50, 49) | 1.88 (1.58, 45) | 2.10 (1.77, 46) | 2.27 (1.43, 47) |
| Estimated mean change from baseline in pain score (95% CI)3 | -1.06 (-1.74, -0.37) | -2.70 (-3.38, -2.02) | -3.11 (-3.81, -2.41) | -3.85 (-4.47, -3.23) | -3.68 (-4.34, -3.01) | -0.44 (-1.14, 0.27) | -1.61 (-2.35, -0.88) | -1.91 (-2.64, -1.18) | -2.55 (-3.25, -1.84) | -2.27 (-3.00, -1.55) |
| Estimated mean ratio to baseline in uterine volume (95% CI) | 1.02 (0.91, 1.15) | 0.83 (0.74, 0.94) | 1.06 (0.94, 1.20) | 0.69 (0.62, 0.77) | 0.92 (0.82, 1.03) | 1.04 (0.92, 1.17) | 0.85 (0.75, 0.96) | 0.88 (0.77, 0.99) | 0.57 (0.50, 0.64) | 0.80 (0.71, 0.91) |
| Estimated mean ratio to baseline in fibroid volume (95% CI) | 0.95 (0.75, 1.19) | 0.75 (0.60, 0.94) | 0.98 (0.77, 1.24) | 0.57 (0.46, 0.70) | 0.88 (0.70, 1.09) | 1.04 (0.84, 1.29) | 0.85 (0.68, 1.06) | 0.93 (0.75, 1.17) | 0.51 (0.41, 0.63) | 0.79 (0.63, 0.99) |
| Estimated mean change from baseline in HRQL score (95% CI)4 | 15.5 (9.4, 21.6) | 26.1 (20.0, 32.2) | 37.2 (31.0, 43.5) | 35.5 (29.8, 41.1) | 34.2 (28.3, 40.1) | 10.3 (4.0, 16.6) | 20.6 (14.1, 27.2) | 22.9 (16.4, 29.5) | 30.2 (23.9, 36.5) | 30.7 (24.2, 37.1) |
1 Amenorrhea was defined as no menstrual blood detected by the alkaline hematin method (not including spotting or MBL <1 to 3 mL) for 35 days and until the end of the treatment up to 24 weeks
2 In women with baseline anaemia (haemoglobin <12 g/dL). n represents the number of women with non-missing data at 24 weeks
3 Pain was assessed using a 0 to 10 numerical rating scale (NRS).
4 The Health-Related Quality of Life (HRQL) score is a part of the validated Uterine Fibroid Symptoms – Quality of Life (UFS-QoL) questionnaire. The score is from 0 to 100 with a higher score indicating better health-related quality of life. The baseline score was about 40.
ABT estradiol 1 mg/norethisterone acetate 0.5 mg; SD standard Deviation; CI confidence interval
Bone mineral density
BMD was assessed using DXA scan at baseline, during treatment (Weeks 24 and 52) and 6 months after the end of treatment (week 76). Subjects at significant risk of osteoporosis, with a history of or known osteoporosis or other metabolic bone disease were excluded from PRIMROSE 1 and PRIMROSE 2 trials.
Mean percentage BMD decreases observed at 24 and 52 weeks were dose- and time-dependent and attenuated by concomitant ABT (Table 5).
At 24 weeks, the change in BMD was most pronounced in women who had full estradiol suppression with Yselty 200 mg (-3.70%). This regimen was not continued for more than 6 months (see section 4.2). The changes were less pronounced in women who received other regimens: -1.99% with Yselty 100 mg, -0.96% Yselty 100 mg with ABT and -1.13% with Yselty 200 mg with ABT.
At 52 weeks, the mean percentage changes from baseline indicated a reduced rate of BMD loss: -2.36% with Yselty 100 mg, -0.93% with Yselty 100 mg with ABT and -1.61% with Yselty 200 mg with ABT. The level of treatment induced BMD loss in this population considered to be clinically meaningful is not well established, and will depend on the individual woman, but in general BMD losses of approximately 3% or more should be reviewed and monitored carefully. It is important to consider the individual woman's baseline BMD, age and overall osteoporosis risk profile when assessing an individual woman's BMD loss, and the benefit-risk of continuing treatment.
At 24 weeks after stopping treatment, most patients had full or partial recovery of lumbar spine BMD: 53%, 52% and 64% for Yselty 100 mg, 100 mg with ABT and 200 mg with ABT, respectively in PRIMROSE 1 and 59%, 80% and 67% for Yselty 100 mg, 100 mg with ABT and 200 mg with ABT in PRIMROSE 2.
The extent and rate of BMD loss when treating women beyond 12 months is currently unknown.
Table 5. Mean percent change from baseline (CfB) in lumbar spine BMD after 24 and 52 weeks of treatment in PRIMROSE 1 and 2:
| Placebo | Yselty 100 mg | Yselty 100 mg+ABT | Yselty 200 mg* | Yselty 200 mg+ABT | |
|---|---|---|---|---|---|
| 24 weeks of treatment | |||||
| Number of subjects | 130 | 121 | 122 | 138 | 127 |
| Mean percent CfB | 0.46 | -1.99 | -0.96 | -3.70 | -1.13 |
| 95% CI | 0.06; 0.85 | -2.47; -1.50 | -1.45; -0.48 | -4.18; -3.22 | -1.60; -0.66 |
| 52 weeks of treatment | |||||
| Number of subjects | 19 | 93 | 84 | - | 97 |
| Mean percent CfB | -0.83** | -2.36 | -0.93 | - | -1.61 |
| 95% CI | -2.08; 0.42 | -3.10; -1.63 | -1.40; -0.47 | - | -2.22; -0.99 |
* Yselty 200 mg was studied up to 6 months.
** Placebo was used up to 12 months in PRIMROSE 1.
Endometriosis-treated population
The efficacy of Yselty was evaluated in one phase 3, randomised, double-blind and placebo-controlled study, Edelweiss 3, including 484 women treated for up to 6 months. Amongst them, 356 women continued into the Edelweiss 6 extension study for an additional 6 months of treatment. A 6-month drug-free post-treatment follow-up evaluated the persistence of efficacy under long-term treatment. This study was conducted primarily in Europe with about 10% of subjects being from US.
Eligible patients consisted in premenopausal women, aged 18 to 49 (inclusive) with surgical confirmed pelvic endometriosis and with moderate to severe endometriosis associated pain (EAP).
Women had a mean age of 34.9 years and mean body mass index was 24.27 kg/m² (range 17.4 to 52.8). Approximately 98.6% of participating women were White.
The mean (SD) time since medical diagnosis of endometriosis was 5.20 (4.24) years. The most commonly reported symptoms, besides pelvic pain, were dyspareunia (88.0%), dyschezia (51.0%) and dysuria (26%). At baseline, 30% of patients presented adenomyosis and 18.2% rectovaginal endometriosis nodes.
The majority of the study population of EDELWEISS 3 reported having undergone previous surgeries/procedures for endometriosis treatment before inclusion into the EDELWEISS studies. Previous medical treatments consisted of analgesics for pelvic pain, including opioids. The most frequently reported other pharmacotherapies for endometriosis treatment included dienogest, hormonal oral contraceptives and GnRH agonists.
Subjects were randomised to one of 3 treatments: placebo (N=162), Yselty 75 mg (N=160), Yselty 200 mg with concomitant ABT (estradiol 1 mg/norethisterone acetate 0.5 mg, referred to as "with ABT") (N=162), all taken once daily. Subjects starting the extension study for additional 6 months stay on the same treatment regimen for 75mg and 200mg with ABT groups but subjects randomised to placebo were re-randomised 1/1 to Yselty 75 mg or Yselty 200 mg with ABT. Subsequently, all subjects underwent additional 6-month post-treatment drug-free period of follow-up to evaluate the persistence of efficacy under long-term treatment.
The two co-primary efficacy endpoints were clinically meaningful reduction of dysmenorrhea (DYS) and non menstrual pelvic pain (NMPP) over the last 28 days of randomized treatment up to the Month 3 visit along with a stable or decreased use of analgesics. These were defined as a reduction of 1.10 or greater from baseline pain for dysmenorrhea and a reduction of 0.80 or greater from baseline pain for non-menstrual pelvic pain, both measured on a 0 (no pain) to 3 (severe pain) verbal rating scale (VRS) using an electronic diary.
Treatment with Yselty 200 mg with ABT demonstrated statistically significant reductions in both co-primary endpoints of DYS and NMPP with a stable or decreased use of analgesics (see Table 6).
Table 6. Reduction of DYS and NMPP (VRS) at Months 3, 6 and 12 – responder analysis (Edelweiss 3, FAS and Edelweiss 6, TEAS):
| Study | EDELWEISS 3 | EDELWEISS 6 | |||
|---|---|---|---|---|---|
| Month 3 | Month 6 | Month 12 | |||
| Treatment | Placebo | LGX 200 mg + ABT | Placebo | LGX 200 mg + ABT | LGX 200 mg + ABT |
| Nobs | 159 | 156 | 115 | 122 | 111 |
| Responders for DYS | |||||
| Percentage of responders | 23.5 | 72.9 | 23.5 | 80.0 | 91.0 |
| OR vs placebo | - | 8.80 | - | 12.98 | - |
| 97.5% CI | - | 4.86; 15.91 | - | 7.00; 24.06 | - |
| Responders for NMPP* | |||||
| Percentage of responders | 30.9 | 47.3 | 38.5 | 57.1 | 67.6 |
| OR vs placebo | - | 2.01 | - | 2.13 | - |
| 97.5% CI | - | 1.18; 3.42 | - | 1.26; 3.60 | - |
ABT = add-back therapy; DYS = dysmenorrhea;; LGX = linzagolix; NMPP = non-menstrual pelvic pain; VRS = verbal rating scale; Nobs = patients with observed data at this timepoint; OR = Odds Ratio; CI = Confidence Interval.
* Reduction of 1.1 (resp. 0.8) for DYS (resp. NMPP) in mean pelvic pain score within last 28 days prior to Month 3 or discontinuation, and stable or decreased use of analgesics for endometriosis within the same calendar days.
Statistically significant reductions were observed in the following secondary endpoints at 6 months in the LGX 200 mg+ABT group compared to placebo: DYS (VRS), NMPP (VRS), dyschezia (NRS), overall pelvic pain, OPP (NRS), and the ability to do daily activities measured using the pain dimension of EHP-30, see Table 7.
Table 7. Summary of analyses of secondary endpoints at Month 6:
| Endpoints | Placebo (N=162) | LGX 200 mg + ABT (N=162) | |
|---|---|---|---|
| LSM (95% CI) | LSM (95% CI) | Diff with PBO (97.5% CI) | |
| CfB in DYS (VRS) | -0.66 (-0.79; -0.53) | -1.83 (-1.96; -1.70) | -1.17 (-1.38; -0.97) |
| CfB in NMPP (VRS) | -0.66 (-0.77; -0.56) | -0.92 (-1.03; -0.82) | -0.26 (-0.43; -0.09) |
| CfB in dyschezia (NRS) | -1.41 (-1.71; -1.12) | -1.99 (-2.29; -1.70) | -0.58 (-1.05; -0.11) |
| CfB in OPP (NRS) | -2.19 (-2.55; -1.84) | -3.39 (-3.74; -3.03) | -1.19 (-1.77; -0.62) |
| CfB in EHP-30 pain dimension | -19.47 (-22.66; -16.28) | -35.60 (-38.73; -32.48) | -16.13 (-21.24; -11.02) |
CfB = change from baseline; DYS = dysmenorrhoea; NMPP = non-menstrual pelvic pain; NRS = numeric rating scale; OPP = overall pelvic pain; VRS = verbal rating; EHP-30 = Endometriosis Health Profile-30; LGX = linzagolix; LSM = least square mean
Scores were computed as mean of daily assessments on the last 28 days prior to Month 6 or discontinuation.
Persistence of efficacy was assessed in the linzagolix 200 mg with ABT group as these subjects continued on the same dosing regimen between Month 6 and Month 12.
Bone mineral density
BMD was assessed using DXA scan at baseline, during treatment (Months 6 and 12) and 6 months after the end of treatment. Subjects at significant risk of osteoporosis, with a history of, or known osteoporosis or other metabolic bone disease were excluded from these trials.
For the recommended dosing regimen linzagolix 200 mg with ABT, the mean percent change in lumbar spine BMD from baseline at Month 6 was -0.79%. Of the subjects with available DXA readings at Baseline and at Month 12, the mean percent change corresponding to this period was -1.10% (Table 8).
Table 8. Mean percent change from baseline (CfB) in lumbar spine BMD at Month 6 and Month 12:
| EDELWEISS 3 and 6 | ||
|---|---|---|
| Placebo N=162 | LGX 200 mg + ABT N=162 | |
| 6 months of treatment | ||
| Number of subjects | 123 | 132 |
| Mean percent CfB | 0.77 | -0.79 |
| 95% CI | 0.40; 1.14 | -1.15; -0.43 |
| 12 months of treatment | ||
| Number of subjects | - | 86 |
| Mean percent CfB | - | -1.10 |
| 95% CI | - | -1.79; -0.41 |
LGX = linzagolix
CfB = change from baseline
Effects on endometrium
Endometrial biopsies were performed in a subset of patients at baseline, week 24 and week 52 as part of the safety assessment in phase 3 studies. Results did not raise any safety concerns.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Yselty in all subsets of the paediatric population in treatment of endometriosis and leiomyoma of uterus (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Following oral administration of a single dose of 100 mg or 200 mg, linzagolix is swiftly absorbed, with Cmax occurring approximately 2 h after administration. Linzagolix shows dose-linear pharmacokinetics and no relevant accumulation at steady state.
Administration of linzagolix (200 mg) with a high fat meal appeared to delay and to slightly decrease peak plasma concentrations, consistent with delayed gastric emptying after the high fat meal but had no effect on the extent of exposure. It is not considered to be of clinical significance.
Distribution
Linzagolix was highly bound (>99%) to plasma proteins, in particular to albumin, and did not partition into red blood cells. The volume of distribution (Vd/F) following 7 consecutive days of oral linzagolix 100 mg or 200 mg administration was 11.067 L (CV: 20.4%) and 11.178 L (CV: 11.8%), respectively.
Biotransformation
Metabolite profiling and identification of linzagolix quantified up to 7 metabolites across plasma, urine, and faeces. The predominant component in the human plasma profiles was unchanged linzagolix. Similarly, linzagolix was the predominant component in urine and one of the major components in faeces. All plasma metabolites were present at less than 10% of the total linzagolix related exposure.
Elimination
Following multiple doses of linzagolix, linzagolix t1/2 was approximately 15 hours. Linzagolix was mainly excreted in urine and approximately one third was eliminated via faeces. Following administration of multiple doses of linzagolix 100 mg and 200 mg, the linzagolix geometric mean apparent clearance (CL/F) was 0.522 L/h (CV: 20.1%) and 0.499 L/h (CV: 15.2%), respectively.
Special populations
The population PK analysis suggests that age does not have a meaningful effect on linzagolix exposure. The analysis showed that Black subjects had a 22.5% decrease in CL/F relative to Caucasian subjects; however, the safety profile of linzagolix between Black and Caucasian subjects was similar.
Based on the population PK analysis, weight was found to influence linzagolix PK. The CL/F in patients weighing 52.7 kg (5th percentile) was predicted to be about 19.2% lower, and in patients weighing 112 kg (95th percentile), about 42% higher than in patients weighing 70 kg. However, subgroup analyses of data from the pivotal phase 3 studies did not indicate any clinically relevant differences with respect to safety and efficacy, and no dose adjustment is recommended.
Hepatic impairment
A clinical study conducted in female subjects with hepatic impairment (mild Child-Pugh A, moderate: Child-Pugh B and severe: Child-Pugh C) revealed no relevant effect on total plasma linzagolix exposure following administration of a single 200 mg dose of linzagolix. The unbound fraction of linzagolix was not affected by mild and moderate hepatic impairment; no dose adjustments with Yselty in patients with mild and moderate hepatic impairment are required (see section 4.2). Yselty should not be used in women with severe hepatic impairment (Child-Pugh C) as 2- to 3-fold higher unbound linzagolix mean exposures were recorded (see section 4.4).
Renal impairment
A clinical study conducted in female subjects with renal impairment (mild, moderate, severe and end-stage renal disease) where glomerular filtration rate (GFR) was assessed using creatine clearance, revealed no relevant effect on total plasma linzagolix exposure following administration of a single 200 mg dose of linzagolix. Unbound plasma linzagolix Cmaxu, AUCu0-t, and AUCu0-inf were increased by 30%, 32%, and 33%, in women with mild renal impairment as compared to healthy subjects with normal renal function. As a potential safety concern with long-term use cannot be excluded, prescribers are recommended to monitor for adverse reactions in women with mild renal impairment (see section 4.4). However, no dose adjustment is required (see section 4.2). Yselty should not be used in women with moderate or severe renal impairment or end-stage renal disease as approximately 1.5-fold (in moderate) and 2-fold (in severe renal impairment and ESRD) higher unbound linzagolix mean exposures were observed (see section 4.4).
5.3. Preclinical safety data
Reproductive and developmental toxicity
Due to its mechanism of action, linzagolix prevented conception and reduced implantation in rat fertility studies and resulted in embryo-foetal mortality, total litter loss or abolished pregnancy in rat and rabbit embryo-foetal studies.
No teratogenic effects and no adverse effect on the pre- and postnatal development were observed in a rat study. Dose levels of 100 mg/kg and 3 mg/kg linzagolix were shown to be the No observed adverse effect level (NOAEL) for reproductive function and embryo-foetal development in the main embryodevelopment studies in rat and rabbit, respectively (corresponding to respectively 5.9 and 0.004 times the maximum recommended human dose based on AUC).
Lactation
Linzagolix was shown to be excreted in milk of rats. Up to 96 h after administration, the radioactivity concentration was lower in milk than in plasma (less than 0.3 times).
Mutagenicity
A standard battery of in vitro and in vivo tests revealed no evidence of mutagenic or clinically relevant genotoxic potential of the drug.
Carcinogenicity
Carcinogenic properties of linzagolix were assessed in a 26-week carcinogenicity study in transgenic Tg RasH2 mice. There was no evidence of linzagolix-induced carcinogenicity up to the highest dose of 500 mg/kg (corresponding to 13.2 times the maximum recommended dose in humans based on AUC).
In a 2-year carcinogenicity study in rats, an increased incidence of uterine endometrial adenocarcinoma was observed in the mid- (50 mg/kg) and high-dose (500 mg/kg) groups (corresponding to respectively 6.8 and 9.6 times the maximum recommended human dose based on AUC) and a marginal increase in the frequency of mammary gland adenocarcinoma was observed at the mid-dose (50 mg/kg) only (6.8 times the maximum recommended human dose based on AUC). The clinical relevance of these findings remains unknown.
Non-carcinogenic histopathological findings in the ovary and uterus (mouse) or ovary and female mammary gland (rat) were considered to be related to the pharmacological action of linzagolix.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.