ZTALMY Oral suspension Ref.[51076] Active ingredients: Ganaxolone
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Marinus Pharmaceuticals Emerald Limited, 10 Earlsfort Terrace, Dublin 2, D02 T380, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antiepileptics, other antiepileptics
ATC code: N03AX27
Mechanism of action
Ganaxolone is a methyl analogue of the endogenous neurosteroid allopregnanolone. Ganaxolone is a neuroactive steroid that positively and allosterically modulates gamma-aminobutyric acid type A (GABAA) receptors in the CNS by interacting with a recognition site that is distinct from other allosteric GABAA receptor modulators.
The precise mechanism by which ganaxolone exerts its therapeutic effects in the treatment of seizures associated with CDD is unknown, but its anticonvulsant effects are thought to result from this modulation of GABAA receptor function providing constant, or tonic, modulation of GABA-mediated inhibitory neurotransmission.
Clinical efficacy and safety
The efficacy to treat seizures associated with CDD in patients 2 years and older was established in a single, double-blind, randomised, placebo-controlled study in patients aged 2 to 19 years (Study 1042-CDD-3001).
Patients enrolled in Study 1042-CDD-3001 had a molecular confirmation of pathogenic or likely pathogenic CDKL5 variant, their seizures were inadequately controlled by at least 2 previous concomitant AED medicinal products, and they had a minimum of 16 seizures of primary seizure type per 28 days in each 1-month period during the 2-month period prior to screening.
Totally, 101 patients were enrolled into the study (51 placebo and 50 study drug). Patients were mostly female (79.2%; consistent with the demographics of CDD) and aged between 2 and 19 years (mean [standard deviation (SD)]: 7.26 [4.55]) with the majority being paediatric (children 2 to 11 years [82.2%], adolescents [16.8%]), concomitant AEDs s were given to 96% patients. The mean (SD) number of concomitant AEDs used by subjects was 2.2 (1.14) in the placebo group and 2.6 (1.40) in the ganaxolone group. The most frequent (≥10 patients) concomitant AEDs were valproate, levetiracetam, clobazam and vigabatrin.
The primary efficacy endpoint was the percentage change from baseline in 28-day frequency of major motor seizures during the 17-week double blind treatment phase. Major motor seizures include bilateral tonic, bilateral clonic, atonic, generalized tonic-clonic and focal to bilateral tonic-clonic seizures. At baseline, the mean (SD) number of major motor seizures over 28-days was 104.8 (173.53) for placebo and 117.2 (138.62) for ganaxolone.
At the end of the 13-week maintenance phase, there was a statistically significant difference in the median percent change from baseline in major motor seizure frequency for patients treated with ganaxolone compared to patients receiving placebo (see Table 1).
Table 1. Study 1042-CDD-3001 Change in frequency of major motor seizures per 28 days in the 13-week maintenance phase:
| Placebo | Ganaxolone | |
|---|---|---|
| 28-Day seizure frequency for primary seizure types, N | 51 | 49 |
| 13-Week Maintenance, Median Percent change (SD) | -6.49 (-26.77, 38.46) | -29.39 (-65.78, 1.30) |
| Wilcoxon Test p-value | 0.0097 | |
| Response Rate, N | 50 | 49 |
| n (%) | 6 (12.0) | 15 (30.6) |
| Difference (95% CI) | 18.6 (2.0, 34.9) | |
| p-valuea | 0.0283 |
CI=95% confidence interval.
a Response is defined as at least 50% reduction from baseline in 28-day Seizure Primary Seizures Frequency. P-value is based on Fisher’s Exact test.
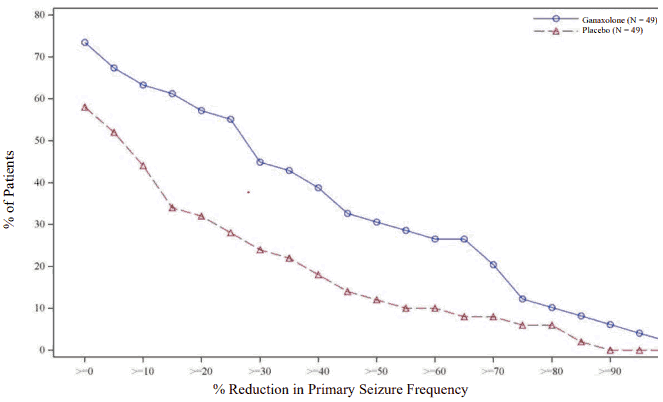
The cumulative response curve shows that ganaxolone produced greater reductions than placebo in seizure frequency at all response rates (see Figure 1).
Figure 1. Study 1042-CDD-3001 Cumulative Responder Curves of 28-Day Seizure Frequency for Primary Seizure Types – 13-Week Maintenance Phase, Intent-to-Treat Population:
Open-label data
CDD patients who participated in the double-blind phase of 1042-CDD-3001 could continue the study and participate in an open-label extension phase. The primary objective of the open-label extension phase was long-term safety and tolerability of ganaxolone. To enter the open-label extension phase, patients underwent a blinded cross-titration to a maximum daily dose of 63 mg/kg/day in patients <28 kg or 1800 mg/day in patients who were at least 28 kg. Data are reported for 88 patients who participated in the open-label extension phase and received ganaxolone for up to 3.5 years. A total of 47.7% of patients discontinued study participation during the open-label extension phase, predominantly due to withdrawal by subject/parent (17.0%), lack of efficacy (15.9%) and adverse events (11.4%).
Adult population
The CDD population in Study 1042-CDD-3001 predominantly consisted of paediatric patients. Two patients were 19 years old at the time of study enrolment (one randomised to placebo, one to ganaxolone). Seven patients turned 18 years of age during the open-label extension phase of the study. For these patients (n=9), the median percent change in major motor seizure frequency from baseline to their last 3 months in the open-label phase was -32.1% (range -86.2% to 72.7%).
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with ZTALMY in one or more subsets of the paediatric population in CDKL5 deficiency disorder (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Ganaxolone is rapidly absorbed, with a time to maximum observed plasma concentration (Tmax) of 2.0 to 3.0 hours at steady state (Css). Css is achieved within 2 to 3 days. Ganaxolone undergoes first-pass metabolism, the absolute bioavailability of ganaxolone suspension is approximately 13%.
Paediatric patients aged 2 to <6 years (median body weight 14.8 kg), aged 6 to <12 years (median body weight 22.6 kg), and aged 12 to <18 years (median body weight 36.1 kg) had a Cmax of 247, 269, and 293 ng/mL and AUC0-24 of 3903, 3998, and 4106 ng*h/mL, respectively, when given a dose of 21 mg/kg with a maximum of 600 mg three times a day. Cmax and AUC0-24 in adult patients was 292 ng/mL and 4100 ng*h/mL, respectively.
Co-administration of ganaxolone with a high-fat meal increased Cmax by 2-fold and AUC by 3-fold when compared to fasted levels. The effect of different types of food is not known.
Distribution
Ganaxolone is extensively distributed throughout the body and its volume of distribution is approximately 580 L. Ganaxolone is approximately 99% protein bound in serum.
Biotransformation
Ganaxolone is extensively metabolized in humans, and over 50 Phase 1 and Phase 2 metabolites have been detected. The ganaxolone metabolite pattern at steady state has not yet been characterised. The steady state metabolite pattern may be different from single dose given the long t1/2 of ganaxolone. Ganaxolone is metabolised by CYP3A4 and CYP3A5; CYP2B6, CYP2C19, CYP2D6, UGT1A3, UGT1A6, UGT1A9, UGT2B7, and UGTB15.
Major metabolite (M2) was identified and demonstrated no activity at the GABAA receptor.
Elimination
The half-life (t½) for ganaxolone at steady state was 7.8 to 10.1 hours. Following a single oral dose of 300 mg [14C]-ganaxolone to healthy male subjects, 55% of the total radioactivity was recovered in feces (2% as unchanged ganaxolone) and 18% of the total radioactivity dose was recovered in urine. Metabolites of ganaxolone may have a longer t½ than ganaxolone, up to 230 hours.
Ganaxolone is excreted in breast milk, concentrations were approximately 4-fold higher than in plasma (see section 4.6).
Dose proportionality and accumulation
The pharmacokinetics of ganaxolone are generally linear between 200 mg and 600 mg (or their paediatric equivalent). When dosing three times a day, Cmax and AUCtau accumulation ratios are 1.5-fold and 1.7-fold, respectively.
Special populations
Effect of age, sex, race
Population pharmacokinetic analyses demonstrated that there were no clinically relevant effects of age, sex, or race on exposure to ganaxolone. CL, V, and maximum absorbed dose all follow an allometric relationship with weight. No clinically relevant effects were observed in children with body weight below 28 kg due to weight-based dosing. Population pharmacokinetic simulations indicate that the ganaxolone exposure in adults was reversely correlated with body weight. The clinical relevance is currently unknown as the efficacy and safety have only been demonstrated for CDD paediatric patients with a low body weight.
Paediatric population
The observed pharmacokinetic exposures in patients in study 1042-CDD-3001 were comparable across the age groups 2 to less than 6 years of age (mean weight 14.8 kg, n=45), 6 to less than 12 years of age (mean weight 22.6 kg, n=28), and 12 to less than 18 years of age (mean weight 36.1 kg, n=16), and greater than 18 years of age (mean weight 35.1 kg, n=2). There are no pharmacokinetic data in children less than 2 years of age.
Renal impairment
The pharmacokinetics of ganaxolone were not significantly altered in patients with severe renal impairment. Following oral administration of a single 300 mg dose in subjects with severe renal impairment (creatinine clearance between 15 and 30 mL/min), the AUC0-inf of ganaxolone decreased 8% and Cmax decreased 11% as compared to that in subjects with normal renal function (creatinine clearance ≥90 mL/min as estimated by Cockcroft-Gault). Patients with end-stage renal disease were not studied.
Hepatic impairment
The influence of hepatic impairment on the pharmacokinetics of ganaxolone was studied following a single oral dose of 300 mg. No clinically significant effects on the exposures of ganaxolone were observed following administration in patients with mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment. Patients with severe (Child-Pugh C) hepatic impairment had an approximately 5.8-fold increase in AUC0-inf as compared to those with normal hepatic function (see section 4.2).
Drug interaction studies
In vitro assessment of drug interactions
In vitro studies with ganaxolone demonstrated that no other pharmacokinetic interactions are expected. Ganaxolone is not an inhibitor or an inducer of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4. In vitro, ganaxolone did not inhibit UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, and UGT2B7. Ganaxolone does not inhibit BCRP, P-gp, MATE1, MATE2-K, OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3 or BSEP. Ganaxolone is not a substrate for BCRP, P-gp, OCT1, OCT2, OATP1B1 or OATP1B3.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology repeated dose toxicity, and genotoxicity.
Repeated dose toxicity
Primary effects in animals were central nervous system clinical observations (e.g., sedation), which were dose-limiting and attributed to exaggerated pharmacology.
In the 12-month repeat-dose toxicology study in dogs, a dose-dependent increase in heart rate at ≥3 mg/kg/day (similar to clinical exposure levels) was observed and there were incidences of sinus tachycardia at higher doses. There were no changes in QTc intervals, blood pressure parameters, or histopathologic correlates.
Carcinogenicity / genotoxicity
Carcinogenicity studies have not been conducted with ganaxolone. Ganaxolone is not considered genotoxic.
Reproductive and developmental toxicity
The reproductive and developmental toxicity studies are of limited value since exposure levels were far below clinically relevant levels.
In the fertility and early embryonic development study in rats, alterations in estrous cyclicity occurred.
In the combined embryo-foetal development and pre- and post-natal development study in rats, gestation length was slightly lengthened and slight delays in offspring growth and related developmental milestones occurred.
Studies in lactating rats indicate that ganaxolone and its metabolites are excreted in milk with concentrations generally higher in milk compared with plasma.
It is not known if ganaxolone crosses the placenta.
Juvenile toxicity
Histological changes in juvenile rats were similar to those in adult rats on an AUC exposure basis. Sedation occurred at lower exposures in adults than in juvenile animals. Decreased bodyweight gain and a delay in sexual maturation occurred in juvenile males and females, with no effects on oestrous cyclicity or any fertility or reproductive parameters. Exposure levels in juvenile animals were similar or lower to the clinical exposure levels.
Ganaxolone administration caused a dose-dependent increase in neurodegeneration in multiple brain regions, consistent with findings from other GABA modulators. There were no functional, neurobehavioural consequences of this effect in the 13-week juvenile study. Exposure levels in juvenile animals were similar or lower to the clinical exposure levels.
Abuse
Ganaxolone shares an internal/subjective interoceptive cue with benzodiazepines and dosedependently supported self-administration in a rodent model of reward, suggesting ganaxolone has reinforcing characteristics similar to benzodiazepines.
Dependence
Animal studies suggest that abrupt discontinuation of ganaxolone may cause withdrawal symptoms.
Studies with metabolites
Based on in vitro data, a potential hormonal effect of metabolite M2 at clinical exposures cannot be excluded. In a 4-week repeat-dose toxicity study with direct administration of M2, acinar atrophy and decreased secretion in the prostate gland and seminal vesicle glands was observed in male rats, which correlated with decreased prostate gland weight. This occurred at levels slightly above clinical exposure levels, and the clinical relevance remains unknown.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.