ZYDELIG Film-coated tablet Ref.[9140] Active ingredients: Idelalisib
Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: Gilead Sciences Ireland UC, Carrigtohill, County Cork, T45 DP77, Ireland
Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Special warnings and precautions for use
Serious infections
Treatment with Zydelig should not be initiated in patients with any evidence of ongoing systemic bacterial, fungal, or viral infection.
Serious and fatal infections have occurred with idelalisib, including opportunistic infections such as Pneumocystis jirovecii pneumonia (PJP) and cytomegalovirus (CMV). Prophylaxis for PJP should therefore be administered to all patients throughout idelalisib treatment, and for a period of 2 to 6 months after discontinuation. The duration of post-treatment prophylaxis should be based on clinical judgment and may take into account a patient’s risk factors such as concomitant corticosteroid treatment and prolonged neutropenia (see section 4.8).
Patients should be monitored for respiratory signs and symptoms throughout treatment. Patients should be advised to report new respiratory symptoms promptly.
Regular clinical and laboratory monitoring for CMV infection is recommended in patients with positive CMV serology at the start of treatment with idelalisib or with other evidence of a history of CMV infection. Patients with CMV viraemia without associated clinical signs of CMV infection should be carefully monitored. For patients with evidence of CMV viraemia and clinical signs of CMV infection, consideration should be given to interrupting idelalisib until the infection has resolved. If the benefits of resuming idelalisib are judged to outweigh the risks, consideration should be given to administering pre-emptive CMV therapy.
Cases of progressive multifocal leukoencephalopathy (PML) have been reported following the use of idelalisib within the context of prior or concomitant immunosuppressive therapies that have been associated with PML. Physicians should consider PML in the differential diagnosis in patients with new or worsening neurological, cognitive or behavioural signs or symptoms. If PML is suspected then appropriate diagnostic evaluations should be undertaken and treatment suspended until PML is excluded. If any doubt exists, referral to a neurologist and appropriate diagnostic measures for PML including MRI scan preferably with contrast, cerebrospinal fluid (CSF) testing for JC viral DNA and repeat neurological assessments should be considered.
Neutropenia
Treatment-emergent Grade 3 or 4 neutropenia, including febrile neutropenia, have occurred in patients treated with idelalisib. Blood counts should be monitored in all patients at least every 2 weeks for the first 6 months of treatment with idelalisib, and at least weekly in patients while ANC is less than 1,000 per mm³ (see section 4.2).
Hepatotoxicity
Elevations in ALT and AST of Grade 3 and 4 (>5 x ULN) have been observed in clinical studies of idelalisib. There have also been reports of hepatocellular injury including hepatic failure. Increases in liver transaminases were generally observed within the first 12 weeks of treatment, and were reversible with dose interruption (see section 4.2). In patients who resumed idelalisib at a lower dose, 26% had recurrence of ALT/AST elevation. Treatment with Zydelig must be withheld in the event of Grade 3 or 4 ALT/AST elevation and liver function monitored. Treatment may be resumed at a lower dose once values have returned to Grade 1 or below (ALT/AST ≤3 x ULN).
ALT, AST, and total bilirubin must be monitored in all patients every 2 weeks for the first 3 months of treatment, then as clinically indicated. If Grade 2 or higher elevations in ALT and/or AST are observed, patients' ALT, AST, and total bilirubin must be monitored weekly until the values return to Grade 1 or below.
Diarrhoea/colitis
Cases of severe drug-related colitis occurred relatively late (months) after the start of therapy, sometimes with rapid aggravation, but resolved within a few weeks with dose interruption and additional symptomatic treatment (e.g., anti-inflammatory agents such as enteric budesonide).
There is very limited experience from the treatment of patients with a history of inflammatory bowel disease.
Pneumonitis and organising pneumonia
Cases of pneumonitis and organising pneumonia (some with fatal outcome) have been reported with idelalisib. In patients presenting with serious lung events, idelalisib should be interrupted and the patient assessed for an explanatory aetiology. If either moderate or severe symptomatic pneumonitis or organising pneumonia is diagnosed, appropriate treatment should be initiated and idelalisib must be permanently discontinued.
Stevens-Johnson syndrome and toxic epidermal necrolysis
Cases of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) with fatal outcomes have been reported when idelalisib was administered concomitantly with other medicinal products associated with these syndromes. If SJS or TEN is suspected, idelalisib should be immediately interrupted and the patient treated accordingly.
CYP3A inducers
Idelalisib exposure may be reduced when co-administered with CYP3A inducers such as rifampicin, phenytoin, St. John’s wort (Hypericum perforatum), or carbamazepine. Since a reduction in idelalisib plasma concentrations may result in decreased efficacy, co-administration of Zydelig with moderate or strong CYP3A inducers should be avoided (see section 4.5).
CYP3A substrates
The primary metabolite of idelalisib, GS-563117, is a strong CYP3A4 inhibitor. Thus, idelalisib has the potential to interact with medicinal products that are metabolised by CYP3A, which may lead to increased serum concentrations of the other product (see section 4.5). When idelalisib is co-administered with other medicinal products, the Summary of Product Characteristics (SmPC) for the other product must be consulted for the recommendations regarding co-administration with CYP3A4 inhibitors. Concomitant treatment of idelalisib with CYP3A substrates with serious and/or life-threatening adverse reactions (e.g., alfuzosin, amiodarone, cisapride, pimozide, quinidine, ergotamine, dihydroergotamine, quetiapine, lovastatin, simvastatin, sildenafil, midazolam, triazolam) should be avoided and alternative medicinal products that are less sensitive to CYP3A4 inhibition should be used if possible.
Hepatic impairment
Intensified monitoring of adverse reactions is recommended in patients with impaired hepatic function as exposure is expected to be increased in this population, in particular in patients with severe hepatic impairment. No patients with severe hepatic impairment were included in clinical studies of idelalisib. Caution is recommended when administering Zydelig in this population.
Chronic hepatitis
Idelalisib has not been studied in patients with chronic active hepatitis including viral hepatitis. Caution should be exercised when administering Zydelig in patients with active hepatitis.
Women of childbearing potential
Women of childbearing potential must use highly effective contraception while taking idelalisib and for 1 month after stopping treatment (see section 4.6). Women using hormonal contraceptives should add a barrier method as a second form of contraception since it is currently unknown whether idelalisib may reduce the effectiveness of hormonal contraceptives.
Excipients
Zydelig contains the azo colouring agent sunset yellow FCF (E110), which may cause allergic reactions.
Interaction with other medicinal products and other forms of interaction
Idelalisib is metabolised primarily via aldehyde oxidase, and to a lesser extent via CYP3A and glucuronidation (UGT1A4). Its primary metabolite is GS-563117, which is not pharmacologically active. Idelalisib and GS-563117 are substrates of P-gp and BCRP.
Effect of other medicinal products on idelalisib pharmacokinetics
CYP3A inducers
A clinical drug interaction study found that co-administration of a single dose of 150 mg idelalisib with rifampicin (a strong CYP3A inducer) resulted in a ~75% reduction in idelalisib AUCinf. Co-administration of Zydelig with moderate or strong CYP3A inducers such as rifampicin, phenytoin, St. John’s wort, or carbamazepine should be avoided as this may result in decreased efficacy (see section 4.4).
CYP3A/P-gp inhibitors
A clinical drug interaction study found that co-administration of a single dose of 400 mg idelalisib with 400 mg once daily ketoconazole (a strong CYP3A, P-gp and BCRP inhibitor) resulted in a 26% increase in Cmax and a 79% increase in AUC inf of idelalisib. No initial dose adjustment of idelalisib is considered necessary when administered with CYP3A/P-gp inhibitors, but an intensified monitoring of adverse reactions is recommended.
Effect of idelalisib on the pharmacokinetics of other medicinal products
CYP3A substrates
The primary metabolite of idelalisib, GS-563117, is a strong CYP3A inhibitor. A clinical drug interaction study found that co-administration of idelalisib with midazolam (a sensitive CYP3A substrate) resulted in a ~140% increase in Cmax and a ~440% increase in AUC inf of midazolam due to the CYP3A inhibition by GS-563117. Co-administration of idelalisib with CYP3A substrates may increase their systemic exposures and increase or prolong their therapeutic activity and adverse reactions. In vitro, the CYP3A4 inhibition was irreversible, and return to normal enzyme activity is therefore expected to take several days after stopping idelalisib administration.
Potential interactions between idelalisib and co-administered medicinal products that are CYP3A substrates are listed in Table 1 (increase is indicated as “↑”). This list is not exhaustive and is intended to serve as guidance only. In general, the SmPC for the other product must be consulted for the recommendations regarding co-administration with CYP3A4 inhibitors (see section 4.4).
Table 1. Interactions between idelalisib and other medicinal products that are CYP3A substrates:
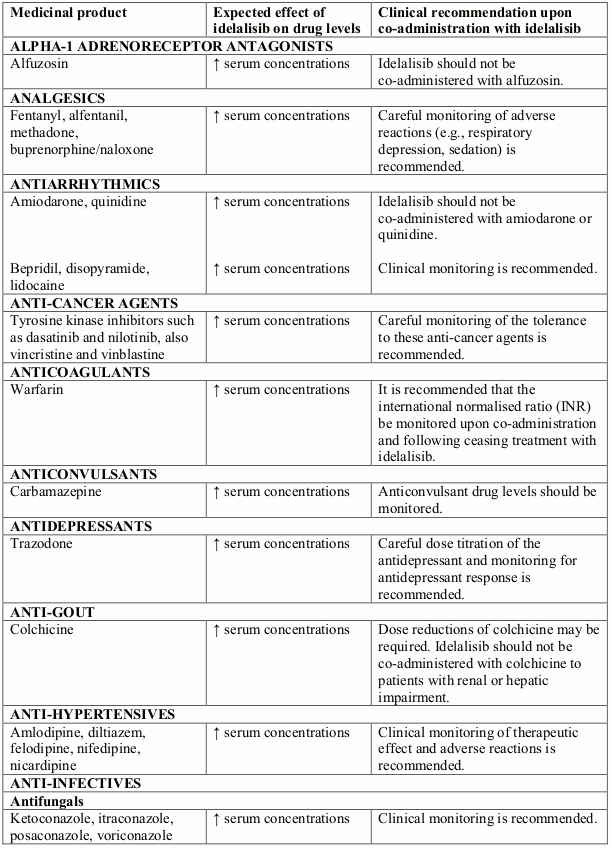
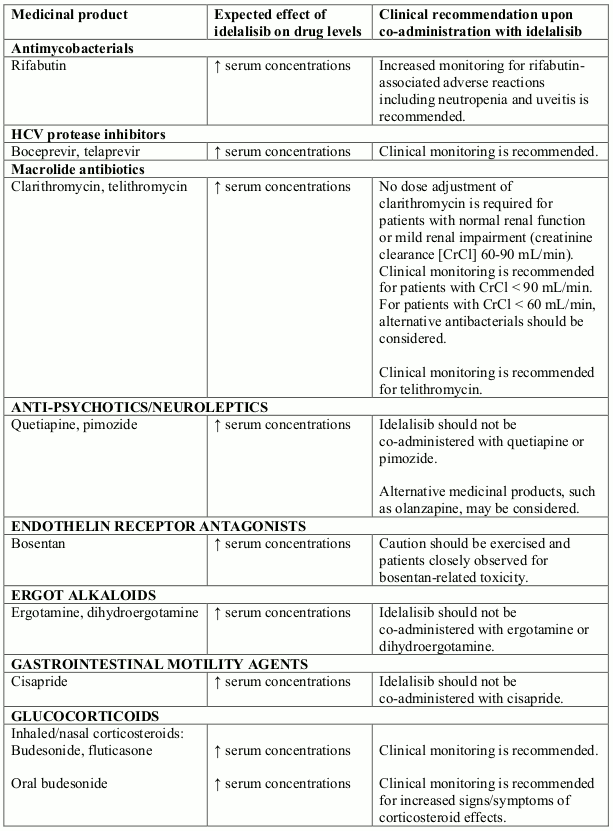

CYP2C8 substrates
In vitro, idelalisib both inhibited and induced CYP2C8, but it is not known whether this translates to an in vivo effect on CYP2C8 substrates. Caution is advised if Zydelig is used together with narrow therapeutic index drugs that are substrates of CYP2C8 (paclitaxel).
Substrates of inducible enzymes (e.g., CYP2C9, CYP2C19, CYP2B6 and UGT)
In vitro, idelalisib was an inducer of several enzymes, and a risk for decreased exposure and thereby decreased efficacy of substrates of inducible enzymes such as CYP2C9, CYP2C19, CYP2B6 and UGT cannot be excluded. Caution is advised if Zydelig is used together with narrow therapeutic index drugs that are substrates of these enzymes (warfarin, phenytoin, S-mephenytoin).
BCRP, OATP1B1, OATP1B3 and P-gp substrates
Co-administration of multiple doses of idelalisib 150 mg twice daily to healthy subjects resulted in comparable exposures for rosuvastatin (AUC 90% CI: 87, 121) and digoxin (AUC 90% CI: 98, 111), suggesting no clinically relevant inhibition of BCRP, OATP1B1/1B3 or systemic P-gp by idelalisib. A risk for P-gp inhibition in the gastrointestinal tract, that could result in increased exposure of sensitive substrates for intestinal P-gp such as dabigatran etexilate, cannot be excluded.
Paediatric population
Interaction studies have only been performed in adults.
Fertility, pregnancy and lactation
Women of childbearing potential / contraception
Based on findings in animals, idelalisib may cause foetal harm. Women should avoid becoming pregnant while taking Zydelig, and for up to 1 month after ending treatment. Therefore, women of childbearing potential must use highly effective contraception while taking Zydelig and for 1 month after stopping treatment. It is currently unknown whether idelalisib may reduce the effectiveness of hormonal contraceptives, and therefore women using hormonal contraceptives should add a barrier method as a second form of contraception.
Pregnancy
There are no or limited amount of data from the use of idelalisib in pregnant women. Studies in animals have shown reproductive toxicity (see section 5.3).
Zydelig is not recommended during pregnancy and in women of childbearing potential not using contraception.
Breast-feeding
It is not known whether idelalisib and its metabolites are excreted in human milk.
A risk to the newborns/infants cannot be excluded.
Breast-feeding should be discontinued during treatment with Zydelig.
Fertility
No human data on the effect of idelalisib on fertility are available. Animal studies indicate the potential for harmful effects of idelalisib on fertility and foetal development (see section 5.3).
Effects on ability to drive and use machines
Zydelig has no or negligible influence on the ability to drive and use machines.
Undesirable effects
Summary of the safety profile
Assessment of adverse reactions is based on two Phase 3 studies (study 312-0116 and study 312-0119) and six Phase 1 and 2 studies. Study 312-0116 was a randomised, double-blind, placebo-controlled study in which 110 subjects with previously treated CLL received idelalisib + rituximab. In addition, 86 subjects from this study who were randomised to receive placebo + rituximab went on to receive idelalisib as a single agent in an extension study (study 312-0117). Study 312-0119 was a randomised, controlled, open-label study in which 173 subjects with previously treated CLL received idelalisib + ofatumumab. The Phase 1 and 2 studies assessed the safety of idelalisib in a total of 536 subjects with haematologic malignancies, including 400 subjects who received idelalisib (any dose) as a single agent and 136 subjects who received idelalisib in combination with an anti-CD20 monoclonal antibody (rituximab or ofatumumab).
Tabulated list of adverse reactions
The adverse drug reactions reported with idelalisib alone or in combination with anti-CD20 monoclonal antibodies (rituximab or ofatumumab) are provided in Table 2. Adverse reactions are listed by system organ class and frequency. Frequencies are defined as follows: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000), and not known (cannot be estimated from available data).
Table 2. Adverse drug reactions reported in clinical studies in subjects with haematologic malignancies receiving idelalisib:
| Reaction | Any grade | Grade ≥3 |
|---|---|---|
| Infections and infestations | ||
| Infections (including Pneumocystis jirovecii pneumonia and CMV)* | Very common | Very common |
| Blood and lymphatic system disorders | ||
| Neutropenia | Very common | Very common |
| Lymphocytosis** | Very common | Very common |
| Respiratory, thoracic and mediastinal disorders | ||
| Pneumonitis | Common | Common |
| Organising pneumonia | Uncommon | Uncommon |
| Gastrointestinal disorders | ||
| Diarrhoea/colitis | Very common | Very common |
| Hepatobiliary disorders | ||
| Transaminase increased | Very common | Very common |
| Hepatocellular injury | Common | Common |
| Skin and subcutaneous tissue disorders | ||
| Rash*** | Very common | Common |
| Stevens-Johnson syndrome/toxic epidermal necrolysis | Rare | Rare |
| General disorders and administration site conditions | ||
| Pyrexia | Very common | Common |
| Investigations | ||
| Increased triglycerides | Very common | Common |
* Comprised of opportunistic infections as well as bacterial and viral infections such as pneumonia, bronchitis, and sepsis.
** Idelalisib-induced lymphocytosis should not be considered progressive disease in the absence of other clinical findings (see section 5.1).
*** Includes the preferred terms dermatitis exfoliative generalised, drug eruption, rash, rash erythematous, rash generalised, rash macular, rash maculo-papular, rash papular, rash pruritic, rash pustular, rash vesicular, papule, skin plaque, and exfoliative rash.
Description of selected adverse reactions
Infections (see section 4.4)
Higher frequencies of infections overall, including Grade 3 and 4 infections, were observed in the idelalisib arms compared to the control arms of idelalisib clinical studies. Most frequently observed were infections in the respiratory system and septic events. In many instances the pathogen was not identified; however, both conventional and opportunistic pathogens, including PJP and CMV, were among those identified. Nearly all PJP infections, including fatal cases, occurred in the absence of PJP prophylaxis. There have been cases of PJP after stopping idelalisib treatment.
Rash
Rash was generally mild to moderate and resulted in discontinuation of treatment in 2.1% of subjects. In studies 312-0116/0117 and 312-0119, rash (reported as dermatitis exfoliative generalised, drug eruption, rash, rash erythematous, rash generalised, rash macular, rash maculo-papular, rash papular, rash pruritic, rash pustular, rash vesicular, papule and skin plaque) occurred in 31.1% of subjects who received idelalisib + an anti-CD20 monoclonal antibody (rituximab or ofatumumab) and 8.2% who received an anti-CD20 monoclonal antibody only (rituximab or ofatumumab). Of these, 5.7% who received idelalisib + an anti-CD20 monoclonal antibody (rituximab or ofatumumab) and 1.5% who received an anti-CD20 monoclonal antibody only (rituximab or ofatumumab) had rash of Grade 3, and no subjects had an adverse reaction of Grade 4. Rash typically resolved with treatment (e.g., topical and/or oral steroids, diphenhydramine) and dose interruption for severe cases (see section 5.3, phototoxicity).
Stevens-Johnson syndrome and toxic epidermal necrolysis (see section 4.4)
Rarely, cases of SJS and TEN have occurred when idelalisib was administered concomitantly with other medicinal products associated with these syndromes (bendamustine, rituximab, allopurinol, and amoxicillin). SJS or TEN occurred within one month of the medicinal combination and fatal outcomes have resulted.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.