Dexlansoprazole
Chemical formula: C₁₆H₁₄F₃N₃O₂S Molecular mass: 369.36 g/mol PubChem compound: 9578005
Mechanism of action
Dexlansoprazole belongs to a class of antisecretory compounds, the substituted benzimidazoles, that suppress gastric acid secretion by specific inhibition of the (H+, K+)-ATPase at the secretory surface of the gastric parietal cell. Because this enzyme is regarded as the acid (proton) pump within the parietal cell, dexlansoprazole has been characterized as a gastric proton-pump inhibitor, in that it blocks the final step of acid production.
Pharmacodynamic properties
Antisecretory Activity
The effects of dexlansoprazole 60 mg (n=20) or lansoprazole 30 mg (n=23) once daily for five days on 24 hour intragastric pH were assessed in healthy subjects in a multiple-dose crossover study. The results are summarized in Table 5.
Effect on 24 Hour Intragastric pH on Day 5 After Administration of Dexlansoprazole or Lansoprazole
| Dexlansoprazole 60 mg | Lansoprazole 30 mg |
|---|---|
| Mean Intragastric pH | |
| 4.55 | 4.13 |
| % Time Intragastric pH >4 (hours) | |
| 71 (17 hours) | 60 (14 hours) |
Serum Gastrin Effects
The effect of dexlansoprazole on serum gastrin concentrations was evaluated in approximately 3460 patients in clinical trials up to eight weeks and in 1023 patients for up to six to 12 months. The mean fasting gastrin concentrations increased from baseline during treatment with 30 and 60 mg dexlansoprazole. In patients treated for more than six months, mean serum gastrin levels increased during approximately the first three months of treatment and were stable for the remainder of treatment. Mean serum gastrin levels returned to pretreatment levels within one month of discontinuation of treatment.
Increased gastrin causes enterochromaffin-like cell hyperplasia and increased serum CgA levels. The increased CgA levels may cause false positive results in diagnostic investigations for neuroendocrine tumors.
Enterochromaffin-Like Cell (ECL) Effects
There were no reports of ECL cell hyperplasia in gastric biopsy specimens obtained from 653 patients treated with dexlansoprazole 30, 60, or 90 mg for up to 12 months.
During lifetime exposure of rats dosed daily with up to 150 mg/kg/day of lansoprazole, marked hypergastrinemia was observed followed by ECL cell proliferation and formation of carcinoid tumors, especially in female rats.
Cardiac Electrophysiology
At a dose five times the maximum recommended dose, dexlansoprazole does not prolong the QT interval to any clinically relevant extent.
Pharmacokinetic properties
12.3 Pharmacokinetics
The dual delayed-release formulation of dexlansoprazole results in a dexlansoprazole plasma concentration-time profile with two distinct peaks; the first peak occurs one to two hours after administration, followed by a second peak within four to five hours (see Figure 1). Dexlansoprazole is eliminated with a half-life of approximately one to two hours in healthy subjects and in patients with symptomatic GERD. No accumulation of dexlansoprazole occurs after multiple, once daily doses of dexlansoprazole 30 or 60 mg although mean AUCt and Cmax values of dexlansoprazole were slightly higher (less than 10%) on Day 5 than on Day 1.
Figure 1. Mean Plasma Dexlansoprazole Concentration – Time Profile Following Oral Administration of 30 or 60 mg Dexlansoprazole Once Daily for 5 Days in Healthy Adult Subjects:
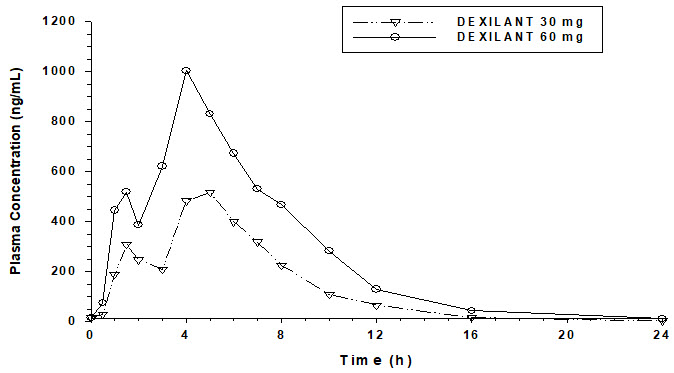
The pharmacokinetics of dexlansoprazole are highly variable, with percent coefficient of variation (CV) values for Cmax, AUC, and CL/F of greater than 30 (see Table 1).
Table 1. Mean (%CV) Pharmacokinetic Parameters for Adult Subjects on Day 5 After Administration of dexlansoprazole:
| Dose (mg) | Cmax (ng/mL) | AUC24 (ng∙h/mL) | CL/F (L/h) |
|---|---|---|---|
| 30 | 658 (40%) (N=44) | 3275 (47%) (N=43) | 11.4 (48%) (N=43) |
| 60 | 1397 (51%) (N=79) | 6529 (60%) (N=73) | 11.6 (46%) (N=41) |
Absorption
After oral administration of dexlansoprazole 30 or 60 mg to healthy subjects and symptomatic GERD patients, mean Cmax and AUC values of dexlansoprazole increased approximately dose proportionally (see Figure 1).
When granules of dexlansoprazole 60 mg are mixed with water and dosed via NG tube or orally via syringe, the bioavailability (Cmax and AUC) of dexlansoprazole was similar to that when dexlansoprazole 60 mg was administered as an intact capsule.
Effect on Food
In food-effect studies in healthy subjects receiving dexlansoprazole under various fed conditions compared to fasting, increases in Cmax ranged from 12 to 55%, increases in AUC ranged from 9 to 37%, and Tmax varied (ranging from a decrease of 0.7 hours to an increase of three hours).
Distribution
Plasma protein binding of dexlansoprazole ranged from 96 to 99% in healthy subjects and was independent of concentration from 0.01 to 20 mcg/mL. The apparent volume of distribution (Vz/F) after multiple doses in symptomatic GERD patients was 40 L.
Elimination
Metabolism
Dexlansoprazole is extensively metabolized in the liver by oxidation, reduction, and subsequent formation of sulfate, glucuronide and glutathione conjugates to inactive metabolites. Oxidative metabolites are formed by the cytochrome P450 (CYP) enzyme system including hydroxylation mainly by CYP2C19, and oxidation to the sulfone by CYP3A4.
CYP2C19 is a polymorphic liver enzyme which exhibits three phenotypes in the metabolism of CYP2C19 substrates: extensive metabolizers (*1/*1), intermediate metabolizers (*1/mutant) and poor metabolizers (mutant/mutant). Dexlansoprazole is the major circulating component in plasma regardless of CYP2C19 metabolizer status. In CYP2C19 intermediate and extensive metabolizers, the major plasma metabolites are 5-hydroxy dexlansoprazole and its glucuronide conjugate, while in CYP2C19 poor metabolizers dexlansoprazole sulfone is the major plasma metabolite.
Excretion
Following the administration of dexlansoprazole, no unchanged dexlansoprazole is excreted in urine. Following the administration of [ 14C] dexlansoprazole to six healthy male subjects, approximately 50.7% (standard deviation (SD): 9.0%) of the administered radioactivity was excreted in urine and 47.6% (SD: 7.3%) in the feces. Apparent clearance (CL/F) in healthy subjects was 11.4 to 11.6 L/hour, respectively, after five days of 30 or 60 mg once daily administration.
Specific Populations
Age: Pediatric Population
The pharmacokinetics of dexlansoprazole in patients under the age of 12 years have not been studied.
Patients 12 to 17 Years of Age:
The pharmacokinetics of dexlansoprazole were studied in 36 patients 12 to 17 years of age with symptomatic GERD in a multicenter trial. Patients were randomized to receive dexlansoprazole 30 or 60 mg once daily for seven days. The dexlansoprazole mean Cmax and AUC in patients 12 to 17 years of age were 105 and 88%, respectively, compared to those observed in adults at the 30 mg dose, and were 81 and 78%, respectively, at the 60 mg dose.
Table 2. Mean (%CV) Pharmacokinetic Parameters in Patients 12 to 17 Years of Age with Symptomatic GERD on Day 7 After Administration of Dexlansoprazole Once Daily for 7 Days:
| Dose | Cmax (ng/mL) | AUCtau (ng∙h/mL) | CL/F (L/h) |
|---|---|---|---|
| 30 mg (N=17) | 691 (53) | 2886 (47) | 12.8 (48) |
| 60 mg (N=18) | 1136 (51) | 5120 (58) | 15.3 (49) |
Age: Geriatric Population
The terminal elimination half-life of dexlansoprazole is significantly increased in geriatric subjects compared to younger subjects (2.2 and 1.5 hours, respectively). Dexlansoprazole exhibited higher systemic exposure (AUC) in geriatric subjects (34% higher) than younger subjects.
Sex
In a study of 12 male and 12 female healthy subjects who received a single dose of dexlansoprazole 60 mg, females had higher systemic exposure (AUC) (43% higher) than males. This difference in exposure between males and females does not represent a significant safety concern.
Renal Impairment
Dexlansoprazole is extensively metabolized in the liver to inactive metabolites, and no parent drug is recovered in the urine following an oral dose of dexlansoprazole. Therefore, the pharmacokinetics of dexlansoprazole are not expected to be altered in patients with renal impairment, and no studies were conducted in patients with renal impairment. In addition, the pharmacokinetics of lansoprazole were not clinically different in patients with mild, moderate or severe renal impairment compared to healthy subjects with normal renal function.
Hepatic Impairment
In a study of 12 patients with moderate hepatic impairment (Child-Pugh Class B) who received a single dose of 60 mg dexlansoprazole, the systemic exposure (AUC) of bound and unbound dexlansoprazole was approximately two times greater compared to subjects with normal hepatic function. This difference in exposure was not due to a difference in protein binding. No studies have been conducted in patients with severe hepatic impairment (Child-Pugh Class C).
Drug-Drug Interactions
Effect of Dexlansoprazole on Other Drugs
Cytochrome P 450 Interactions:
Dexlansoprazole is metabolized, in part, by CYP2C19 and CYP3A4.
In vitro studies have shown that dexlansoprazole is not likely to inhibit CYP isoforms 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 2E1 or 3A4. As such, no clinically relevant interactions with drugs metabolized by these CYP enzymes would be expected. Furthermore, in vivo studies showed that dexlansoprazole did not have an impact on the pharmacokinetics of coadministered phenytoin (CYP2C9 substrate) or theophylline (CYP1A2 substrate).
The subjects' CYP1A2 genotypes in the drug-drug interaction study with theophylline were not determined. Although in vitro studies indicated that dexlansoprazole has the potential to inhibit CYP2C19 in vivo, an in vivo drug-drug interaction study in mainly CYP2C19 extensive and intermediate metabolizers has shown that dexlansoprazole does not affect the pharmacokinetics of diazepam (CYP2C19 substrate).
Clopidogrel:
Clopidogrel is metabolized to its active metabolite in part by CYP2C19. A study of healthy subjects who were CYP2C19 extensive metabolizers, receiving once daily administration of clopidogrel 75 mg alone or concomitantly with dexlansoprazole 60 mg (n=40), for nine days was conducted. The mean AUC of the active metabolite of clopidogrel was reduced by approximately 9% (mean AUC ratio was 91%, with 90% CI of 86 to 97%) when dexlansoprazole was coadministered compared to administration of clopidogrel alone. Pharmacodynamic parameters were also measured and demonstrated that the change in inhibition of platelet aggregation (induced by 5 mcM ADP) was related to the change in the exposure to clopidogrel active metabolite. The effect on exposure to the active metabolite of clopidogrel and on clopidogrel-induced platelet inhibition is not considered clinically important.
Effect of Other Drugs on Dexlansoprazole
Because dexlansoprazole is metabolized by CYP2C19 and CYP3A4, inducers and inhibitors of these enzymes may potentially alter exposure of dexlansoprazole.
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.