ATTRUBY Film-coated tablet Ref.[113517] Active ingredients: Acoramidis
Source: FDA, National Drug Code (US) Revision Year: 2024
12.1. Mechanism of Action
Acoramidis is a selective stabilizer of transthyretin (TTR). Acoramidis binds TTR at thyroxine binding sites and slows dissociation of the TTR tetramer into its constituent monomers, the rate-limiting step in amyloidogenesis.
12.2. Pharmacodynamics
TTR Stabilization
Changes in serum TTR level or in vitro TTR stabilization assays were utilized as pharmacodynamic markers of TTR stabilization. Increases in mean serum TTR levels were observed by Day 28 in ATTR-CM patients treated with ATTRUBY. Near-complete in vitro TTR stabilization was observed as early as Day 28 and through completion of a 30-month study of patients with ATTR-CM (wild-type and variant) treated with the recommended dosage [see Clinical Studies (14)].
Free thyroxine
ATTRUBY may decrease serum concentrations of free thyroxine without an accompanying change in thyroid stimulating hormone (TSH). A reduction in free thyroxine values has been observed with transthyretin stabilizers probably due to reduced thyroxine binding to or displacement from transthyretin (TTR).
NT-proBNP and Troponin I
In a clinical study of ATTRUBY in patients with ATTR-CM, at Month 30, the increase in N-terminal prohormone of brain natriuretic peptide [NT-proBNP] and troponin I was lower with ATTRUBY versus placebo. The increase in NT-proBNP at Month 30 for ATTRUBY was about half that of placebo [see Clinical Studies (14)].
Cardiac Electrophysiology
At approximately 1.2 times the steady state peak plasma concentrations (Cmax) at the recommended dose, ATTRUBY does not prolong the QTc interval to any clinically relevant extent.
12.3. Pharmacokinetics
The systemic exposures (Cmax and AUC) increase in a less than dose proportional manner following single and multiple doses of acoramidis. Over the dose range from 89 mg twice daily to 712 mg twice daily, AUC increases only 130%. Acoramidis steady state is achieved by 4 days with approximately 1.3-fold accumulation at the approved recommended dosage. At steady state, a dose of 712 mg twice daily results in a mean (SD) Cmax of 13700 (6090) ng/mL and AUC0-12h of 47200 (10300) ng.h/mL.
Absorption
The time to Cmax of acoramidis (Tmax) is approximately 1 hour following oral administration.
Effect of Food
No clinically significant differences in acoramidis pharmacokinetics were observed following administration of a high-fat meal (800-1000 total calories, ≥ 50% fat).
Distribution
The apparent steady-state volume of distribution for acoramidis is 654 liters. Acoramidis is 96% bound to human plasma proteins in vitro. Acoramidis primarily binds to TTR.
Elimination
The effective half-life of acoramidis is approximately 6 hours with a steady state apparent clearance of 16 L/hr.
Metabolism
Acoramidis is primarily metabolized by glucuronidation via UGT1A9, UGT1A1 and UGT2B7. Acoramidis-β-D-glucuronide (Acoramidis-AG) is the predominant metabolite of acoramidis (8% of total circulating drug related moieties).
Acoramidis-AG is approximately ⅓ as pharmacologically active compared with acoramidis, has a low potential for covalent binding, and does not contribute to pharmacological activity.
Excretion
After a single oral dose of radiolabeled acoramidis 712 mg to healthy adult subjects, approximately 32% of the dose radioactivity was recovered in feces (15% unchanged), and approximately 68% was recovered in urine (<10% unchanged).
Specific Populations
No clinically significant differences in the pharmacokinetics of acoramidis were observed based on age, race/ethnicity (including Japanese and non-Japanese), sex, or renal impairment. The effect of hepatic impairment (Child Pugh A, B, or C) on acoramidis pharmacokinetics is unknown.
Drug Interaction Studies
Clinical Studies
Following the administration of acoramidis (712 mg, BID) in a clinical study in healthy adult volunteers, there was not a clinically significant increase in exposure to the organic anion transporter-1 (OAT1) substrate (adefovir) and to OAT3 substrate (oseltamivir carboxylate). Concomitant diuretic use in patients does not affect steady-state plasma acoramidis concentrations.
In vitro Studies
Cytochrome P450 Enzymes:
Acoramidis is a time-dependent inhibitor of CYP2C9, but does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C19, CYP2D6, or CYP3A4/5. Acoramidis does not induce CYP1A2, CYP2B6, or CYP3A4.
UDP-Glucuronosyl Transferase (UGT):
Acoramidis is a substrate of multiple UGT enzymes including UGT1A9, UGT1A1, and UGT2B7.
Transporter Systems:
Acoramidis is a substrate for OAT1 and breast cancer resistance protein (BCRP). Acoramidis inhibits OAT1 and OAT3, but does not inhibit MATE1, OCT1, OCT2, OATP1B1, OATP1B3, MATE2-K, BCRP, P-gp, or BSEP.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
There was no evidence of increased incidence of neoplasia in a 2-year carcinogenicity study in male rats dosed up to 50 mg/kg and in female rats dosed up to 350 mg/kg, which provided exposures approximately equivalent to and 11 times the AUC at the maximum recommended human dose (MRHD), respectively. There was no evidence of an increased incidence of neoplasia in transgenic (Tg.rasH2) mice following repeated daily administration for 26 weeks at daily doses up to 300 mg/kg.
Mutagenesis
There was no evidence of mutagenicity or clastogenicity for acoramidis in an Ames assay or in vivo rat micronucleus and alkaline comet assay.
Impairment of Fertility
In a male and female fertility study, rats were orally administered acoramidis at 0, 50, 350, and 1,000 mg/kg/day. Male rats were given acoramidis prior to and during cohabitation for a total of up to 52 days. Female rats were given acoramidis prior to and during cohabitation until implantation of the embryo (Gestational Day 7) for a total of up to 34 days. There were no effects on fertility, reproductive performance, or mating behavior in male or female rats at doses up to 1,000 mg/kg/day, approximately 38-times the AUC at the MRHD.
14. Clinical Studies
The efficacy of ATTRUBY was demonstrated in a multicenter, international, randomized, double-blind, placebo-controlled study in 611 adult patients with wild-type or variant (hereditary or de novo) ATTR-CM (NCT03860935).
Participants were randomized (2:1) to receive ATTRUBY 712 mg (n=409) or placebo (n=202) twice daily for 30 months. Treatment assignment was stratified by type of ATTR-CM [variant (ATTRv-CM) or wild-type (ATTRwt-CM)], NT-proBNP level, and estimated glomerular filtration rate (eGFR). The mean age of study participants was 77 years, 90.8% were male, 87.9% were White, 4.7% Black or African American, 2.1% Asian, 5.3% race other, 19% had a history of permanent pacemaker and 58% had a history of atrial fibrillation. No significant imbalance in baseline characteristics was observed between the two treatment groups.
Participants were permitted to initiate open-label tafamidis after 12 months in the study. A total of 107 participants received tafamidis: 61 (14.9%) in the ATTRUBY arm and 46 (22.8%) in the placebo arm. The median time to initiation of tafamidis for these 107 participants was 17 months.
The primary composite endpoint included all-cause mortality (ACM) and cumulative frequency of cardiovascular-related hospitalizations (CVH) over 30 months, analyzed hierarchically using the stratified Finkelstein-Schoenfeld (F-S) test. The F-S test demonstrated a statistically significant reduction (p=0.018) in ACM and cumulative frequency of CVH in the ATTRUBY arm versus the placebo arm. All-cause mortality was reported in 19% and 26% of participants in the ATTRUBY and placebo groups, respectively. The majority (79%) of the deaths were cardiovascular. CVH was reported in 27% and 43% of participants in the ATTRUBY and placebo groups, respectively. The mean number of CVH events was 0.3 vs 0.6 per year. The majority (59%) of CVH were heart failure hospitalizations reported in 13% and 26% of the participants in the ATTRUBY and placebo groups, respectively.
The treatment effect of ATTRUBY on functional capacity and health status was assessed by the 6MWD and the Kansas City Cardiomyopathy Questionnaire-Overall Summary score (KCCQ-OS) respectively. At month 30, the LS mean difference (95% CI) in change from baseline in 6MWD was 40 [21, 58] meters (p<0.0001) and change from baseline in KCCQ-OS was 10 [6, 14] points (p<0.0001) (Figure 1 and Figure 2).
Figure 1. 6MWD Change from Baseline:
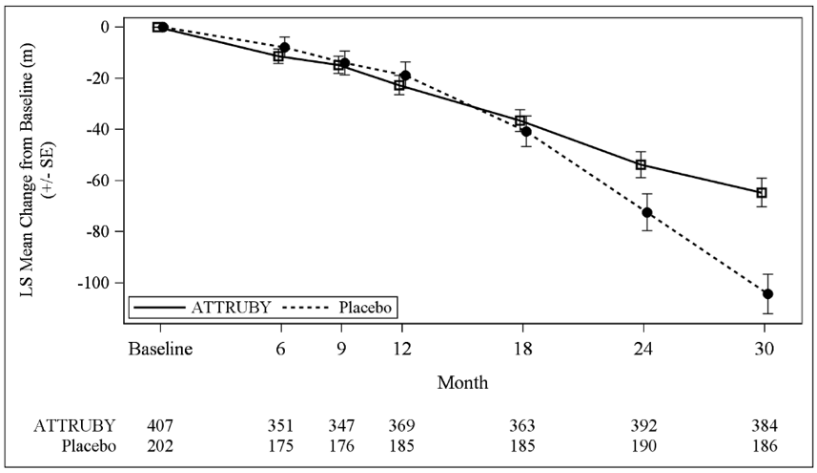
Figure 2. KCCQ-OS Score Change from Baseline:
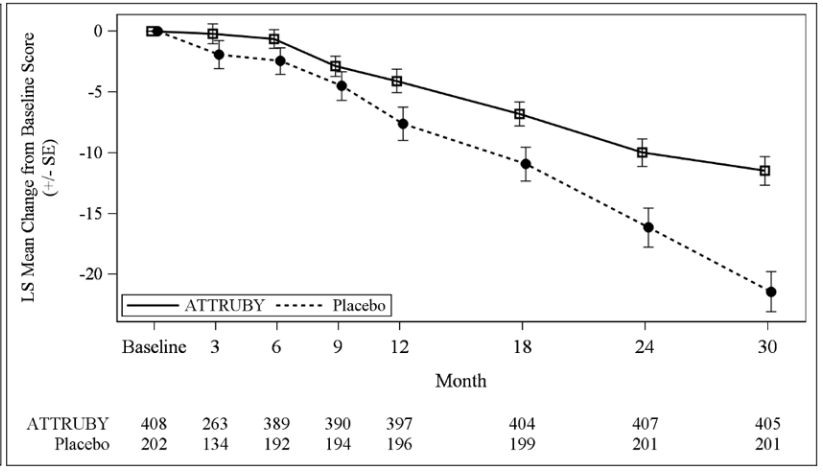
Abbreviations: 6MWD = Six-Minute Walk Distance; KCCQ-OS = Kansas City Cardiomyopathy Questionnaire Overall Summary Score; SE = standard error; LS = least squares.
The changes from baseline in 6MWT and KCCQ-OS were analyzed using the mixed model for repeated measures (MMRM) with treatment group, visit, genotype (ATTRv-CM vs ATTRwt-CM), NT-proBNP level (≤3000 vs >3000 pg/mL), eGFR level (≥45 vs <45 mL/min/1.73 m²) and treatment group-by-visit interaction as factors, and baseline value as covariate.
A Cox regression analysis indicated a 35.5% decrease in the risk of the composite of ACM or first CV hospitalization (hazard ratio: 0.645 [95% CI: 0.500, 0.832]). A Kaplan-Meier plot of time to first event of ACM or CVH is shown in Figure 3.
Figure 3. Time to First All-cause Mortality or Cardiovascular-Related Hospitalization over Month 30, mITT Population:
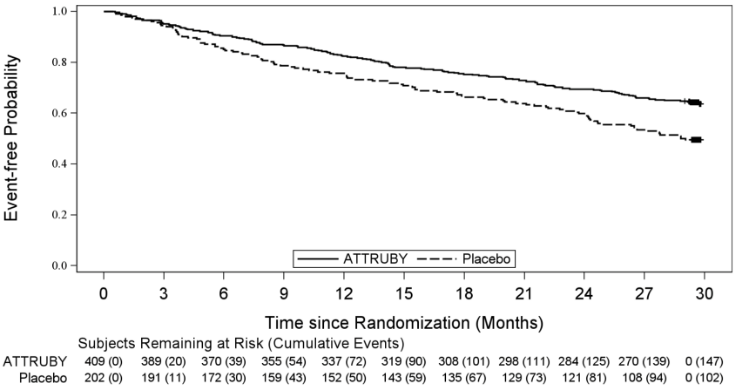
Figure 4 shows the treatment effects by prespecified subgroups.
Figure 4. Win-Ratio Analyses for Hierarchical Combination of All-Cause Mortality and Cardiovascular-Related Hospitalization by Overall and Subgroup, mITT Population:
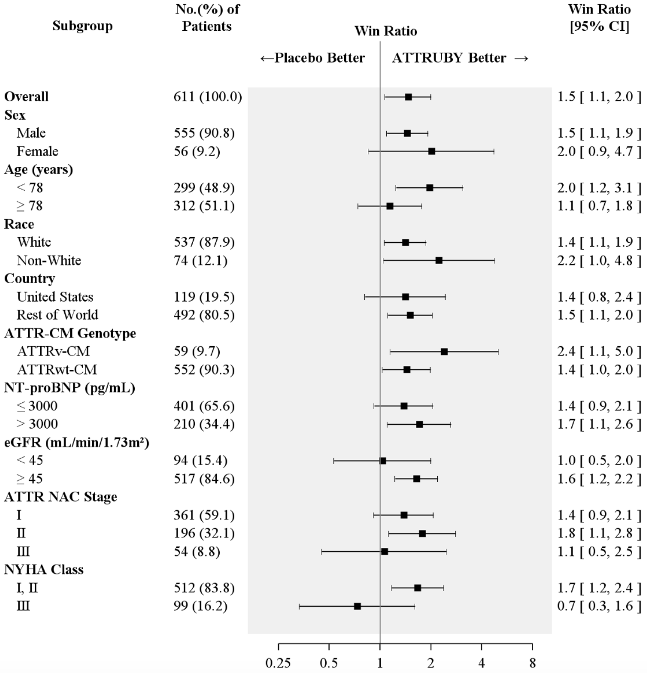
Abbreviations: ATTR-CM = transthyretin amyloid cardiomyopathy; ATTRv-CM = variant ATTR-CM; ATTRwt-CM = wild-type ATTR-CM; eGFR = estimated glomerular filtration rate; mITT=modified intent-to-treat; NAC = National Amyloidosis Centre; NT-proBNP = N-terminal prohormone of brain natriuretic peptide; NYHA = New York Heart Association
All-Cause Mortality includes heart transplant, CMAD and all-cause death. Cardiovascular-related hospitalizations include cardiovascular hospitalizations and urgent unplanned visits requiring treatment with intravenous diuretic for decompensated heart failure. Non-White includes American Indian or Alaska Native, Asian, Black or African American, Native Hawaiian or Other Pacific Islander, Other, multiple races and Not reported.
ATTR NAC Stage: ATTR Stage I, defined as NT-proBNP ≤3000 ng/L and eGFR ≥45 mL/min/1.73 m²; Stage III, defined as NT-proBNP >3000 ng/L and eGFR <45 mL/min/1.73 m², the remainder categorized as Stage II.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.