DACOGEN Powder for concentrate for solution for infusion Ref.[9063] Active ingredients: Decitabine
Source: European Medicines Agency (EU) Revision Year: 2021 Publisher: Janssen-Cilag International NV, Turnhoutseweg 30, B-2340, Beerse, Belgium
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, antimetabolites, pyrimidine analogues
ATC Code: L01BC08
Mechanism of action
Decitabine (5-aza-2'-deoxycytidine) is a cytidine deoxynucleoside analogue that selectively inhibits DNA methyltransferases at low doses, resulting in gene promoter hypomethylation that can result in reactivation of tumour suppressor genes, induction of cellular differentiation or cellular senescence followed by programmed cell death.
Clinical experience
The use of Dacogen was studied in an open-label, randomised, multicentre Phase III study (DACO-016) in subjects with newly diagnosed de novo or secondary AML according to the WHO classification. Dacogen (n=242) was compared to treatment choice (TC, n=243) which consisted of patient’s choice with physician’s advice of either supportive care alone (n=28, 11.5%) or 20 mg/m² cytarabine subcutaneously once daily for 10 consecutive days repeated every 4 weeks (n=215, 88.5%). Dacogen was administered as a 1-hour intravenous infusion of 20 mg/m² once daily for 5 consecutive days repeated every 4 weeks.
Subjects who were considered candidates for standard induction chemotherapy were not included in the study as shown by the following baseline characteristics. The median age for the intent-to-treat (ITT) population was 73 years (range 64 to 91 years). Thirty-six percent of subjects had poor-risk cytogenetics at baseline. The remainder of the subjects had intermediate-risk cytogenetics. Patients with favourable cytogenetics were not included in the study. Twenty-five percent of subjects had an ECOG performance status ≥2. Eighty-one percent of subjects had significant comorbidities (e.g., infection, cardiac impairment, pulmonary impairment). The number of patients treated with Dacogen by racial group was White 209 (86.4%) and Asian 33 (13.6%).
The primary endpoint of the study was overall survival. The secondary endpoint was complete remission rate that was assessed by independent expert review. Progression-free survival and Event-free survival were tertiary endpoints.
The median overall survival in the ITT population was 7.7 months in subjects treated with Dacogen compared to 5.0 months for subjects in the TC arm (hazard ratio 0.85; 95% CI: 0.69, 1.04, p=0.1079). The difference did not reach statistical significance, however, there was a trend for improvement in survival with a 15% reduction in the risk of death for subjects in the Dacogen arm (Figure 1). When censored for potentially disease modifying subsequent therapy (i.e., induction chemotherapy or hypomethylating agent) the analysis for overall survival showed a 20% reduction in the risk of death for subjects in the Dacogen arm [HR=0.80, (95% CI: 0.64, 0.99), p-value = 0.0437)].
Figure 1. Overall survival (ITT population):

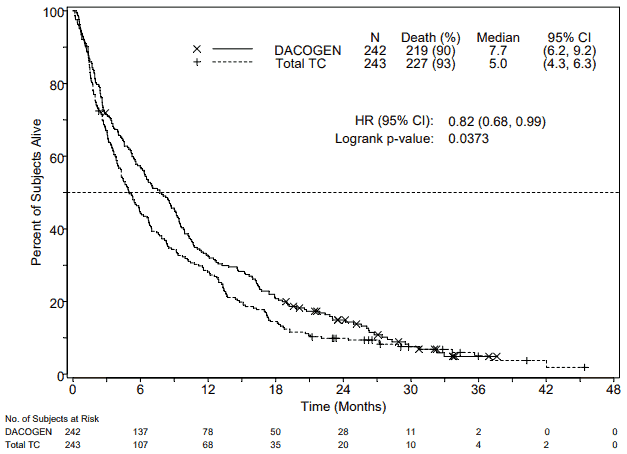
In an analysis with an additional 1 year of mature survival data, the effect of Dacogen on overall survival demonstrated a clinical improvement compared to the TC arm (7.7 months vs. 5.0 months, respectively, hazard ratio = 0.82, 95% CI: 0.68, 0.99, nominal p-value = 0.0373, Figure 2).
Figure 2. Analysis of mature overall survival data (ITT population):
Based on the initial analysis in the ITT population, a statistically significant difference in complete remission rate (CR + CRp) was achieved in favour of subjects in the Dacogen arm, 17.8% (43/242) compared to the TC arm, 7.8% (19/243); treatment difference 9.9% (95% CI: 4.07; 15.83), p=0.0011. The median time to best response and median duration of best response in patients who achieved a CR or CRp were 4.3 months and 8.3 months, respectively. Progression-free survival was significantly longer for subjects in the Dacogen arm, 3.7 months (95% CI: 2.7, 4.6) compared with subjects in the TC arm, 2.1 months (95% CI: 1.9, 3.1); hazard ratio 0.75 (95% CI: 0.62, 0.91), p=0.0031. These results as well as other endpoints are shown in Table 2.
Table 2. Other efficacy endpoints for Study DACO-016 (ITT population):
| Outcomes | Dacogen n=242 | TC (combined group) n=243 | p-value |
|---|---|---|---|
| CR + CRp | 43 (17.8%) | 19 (7.8%) | 0.0011 |
| OR = 2.5 (1.40, 4.78)b | |||
| CR | 38 (15.7%) | 18 (7.4%) | - |
| EFSa | 3.5 (2.5, 4.1)b | 2.1 (1.9, 2.8)b | 0.0025 |
| HR = 0.75 (0.62, 0.90)b | |||
| PFSa | 3.7 (2.7, 4.6)b | 2.1 (1.9, 3.1)b | 0.0031 |
| HR = 0.75 (0.62, 0.91)b | |||
CR = complete remission; CRp = complete remission with incomplete platelet recovery, EFS = event-free survival, PFS = progression-free survival, OR = odds ratio, HR = hazard ratio
- = Not evaluable
a Reported as median months
b 95% confidence intervals
Overall survival and complete remission rates in pre-specified disease-related sub-groups (i.e., cytogenetic risk, Eastern Cooperative Oncology Group [ECOG] score, age, type of AML, and baseline bone marrow blast count) were consistent with results for the overall study population.
The use of Dacogen as initial therapy was also evaluated in an open-label, single-arm, Phase II study (DACO-017) in 55 subjects >60 years with AML according to the WHO classification. The primary endpoint was complete remission (CR) rate that was assessed by independent expert review. The secondary endpoint of the study was overall survival. Dacogen was administered as a 1-hour intravenous infusion of 20 mg/m² once daily for 5 consecutive days repeated every 4 weeks. In the ITT analysis, a CR rate of 23.6% (95% CI: 13.2, 37) was observed in 13/55 subjects treated with Dacogen. The median time to CR was 4.1 months, and the median duration of CR was 18.2 months. The median overall survival in the ITT population was 7.6 months (95% CI: 5.7, 11.5).
The efficacy and safety of Dacogen has not been evaluated in patients with acute promyelocytic leukaemia or CNS leukaemia.
Paediatric population
A Phase I/II open-label, multicentre study evaluated the safety and efficacy of Dacogen in sequential administration with cytarabine in children aged 1 month to <18 years with relapsed or refractory AML. A total of 17 subjects were enrolled and received Dacogen 20 mg/m² in this study, of which 9 subjects received cytarabine 1 g/m² and 8 subjects received cytarabine administered at the maximum tolerable dose of 2 g/m². All subjects discontinued the study treatment. The reasons for treatment discontinuation included disease progression (12 [70.6%] subjects), subjects proceeding to transplant (3 [17.6%]), investigator decision (1 [5.9%]), and “other” (1 [5.9%]). Reported adverse events were consistent with the known safety profile of Dacogen in adults (see section 4.8). Based on these negative results, Dacogen should not be used in children with AML aged <18 years, because efficacy was not established (see section 4.2).
Pharmacokinetic properties
The population pharmacokinetic (PK) parameters of decitabine were pooled from 3 clinical studies in 45 patients with AML or myelodysplastic syndrome (MDS) utilizing the 5-Day regimen. In each study, decitabine PK was evaluated on the fifth day of the first treatment cycle.
Distribution
The pharmacokinetics of decitabine following intravenous administration as a 1-hour infusion were described by a linear two-compartment model, characterised by rapid elimination from the central compartment and by relatively slow distribution from the peripheral compartment. For a typical patient (weight 70 kg/body surface area 1.73 m²) the decitabine pharmacokinetic parameters are listed in the Table 3 below.
Table 3. Summary of population PK analysis for a typical patient receiving daily 1-hour infusions of Dacogen 20 mg/m² over 5 days every 4 weeks:
| Παράμετροςα | Προβλεπόμενη τιμή | 95% ΔΕ |
|---|---|---|
| Cmax (ng/ml) | 107 | 88,5-129 |
| AUCcum (ng.h/ml) | 580 | 480-695 |
| t1/2 (min) | 68,2 | 54,2-79,6 |
| Vdss (L) | 116 | 84,1-153 |
| CL (L/h) | 298 | 249-359 |
a The total dose per cycle was 100 mg/m²
Decitabine exhibits linear PK and following the intravenous infusion, steady-state concentrations are reached within 0.5 hour. Based on model simulation, PK parameters were independent of time (i.e., did not change from cycle to cycle) and no accumulation was observed with this dosing regimen. Plasma protein binding of decitabine is negligible (<1%). Decitabine Vd ss in cancer patients is large indicating distribution into peripheral tissues. There was no evidence of dependencies on age, creatinine clearance, total bilirubin, or disease.
Biotransformation
Intracellularly, decitabine is activated through sequential phosphorylation via phosphokinase activities to the corresponding triphosphate, which is then incorporated by the DNA polymerase. In vitro metabolism data and the human mass balance study results indicated that the cytochrome P450 system is not involved in the metabolism of decitabine. The primary route of metabolism is likely through deamination by cytidine deaminase in the liver, kidney, intestinal epithelium and blood. Results from the human mass-balance study showed that unchanged decitabine in plasma accounted for approximately 2.4% of total radioactivity in plasma. The major circulating metabolites are not believed to be pharmacologically active. The presence of these metabolites in urine together with the high total body clearance and low urinary excretion of unchanged decitabine in the urine (~4% of the dose) indicate that decitabine is appreciably metabolized in vivo. In vitro studies show that decitabine does not inhibit nor induce CYP 450 enzymes up to more than 20-fold of the therapeutic maximum observed plasma concentration (Cmax). Thus; CYP-mediated metabolic drug interactions are not anticipated, and decitabine is unlikely to interact with agents metabolized through these pathways. In addition, in vitro data show that decitabine is a poor P-gp substrate.
Elimination
Mean plasma clearance following intravenous administration in cancer subjects was >200 L/h with moderate inter-subject variability (coefficient of variation [CV] is approximately 50%). Excretion of unchanged drug appears to play only a minor role in the elimination of decitabine.
Results from a mass balance study with radioactive 14 C-decitabine in cancer patients showed that 90% of the administered dose of decitabine (4% unchanged drug) is excreted in the urine.
Additional information on special populations
The effects of renal or hepatic impairment, gender, age or race on the pharmacokinetics of decitabine have not been formally studied. Information on special populations was derived from pharmacokinetic data from the 3 studies noted above, and from one Phase I study in MDS subjects, (N=14; 15 mg/m² x 3-hours q8h x 3 days).
Elderly
Population pharmacokinetic analysis showed that decitabine pharmacokinetics are not dependent on age (range studied 40 to 87 years; median 70 years).
Paediatric population
Population PK analysis of decitabine showed that after accounting for body size, there is no difference between decitabine PK parameters in paediatric AML patients versus adults with AML or MDS.
Gender
Population pharmacokinetic analysis of decitabine did not show any clinically relevant difference between men and women.
Race
Most of the patients studied were Caucasian. However, the population pharmacokinetic analysis of decitabine indicated that race had no apparent effect on the exposure to decitabine.
Hepatic impairment
The PK of decitabine have not been formally studied in patients with hepatic impairment. Results from a human mass-balance study and in vitro experiments mentioned above indicated that the CYP enzymes are unlikely to be involved in the metabolism of decitabine. In addition, the limited data from the population PK analysis indicated no significant PK parameter dependencies on total bilirubin concentration despite a wide range of total bilirubin levels. Thus, decitabine exposure is not likely to be affected in patients with impaired hepatic function.
Renal impairment
The PK of decitabine have not been formally studied in patients with renal insufficiency. The population PK analysis on the limited decitabine data indicated no significant PK parameter dependencies on normalised creatinine clearance, an indicator of renal function. Thus, decitabine exposure is not likely to be affected in patients with impaired renal function.
Preclinical safety data
Formal carcinogenicity studies have not been performed with decitabine. Evidence from the literature indicates that decitabine has carcinogenic potential. The available data from in vitro and in vivo studies provide sufficient evidence that decitabine has genotoxic potential. Data from the literature also indicate that decitabine has adverse effects on all aspects of the reproductive cycle, including fertility, embryo-foetal development and post-natal development. Multi-cycle repeat-dose toxicity studies in rats and rabbits indicated that the primary toxicity was myelosuppression, including effects on bone marrow, which was reversible on cessation of treatment. Gastrointestinal toxicity was also observed and in males, testicular atrophy which did not reverse over the scheduled recovery periods. Decitabine administration to neonatal/juvenile rats showed a comparable general toxicity profile as in older rats. Neurobehavioural development and reproductive capacity were unaffected when neonatal/juvenile rats were treated at dose levels inducing myelosuppression. See section 4.2 for information on paediatric use.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.