DELSTRIGO Film-coated tablet Ref.[28332] Active ingredients: Doravirine Lamivudine Lamivudine, Tenofovir disoproxil and Doravirine Tenofovir disoproxil
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Merck Sharp & Dohme B.V., Waarderweg 39, 2031 BN Haarlem, The Netherlands
4.3. Contraindications
Hypersensitivity to the active substances or to any of the excipients listed in section 6.1.
Co-administration with medicinal products that are strong cytochrome P450 CYP3A enzyme inducers is contraindicated as significant decreases in doravirine plasma concentrations are expected to occur, which may decrease the effectiveness of Delstrigo (see sections 4.4 and 4.5). These medicinal products include, but are not limited to the following:
- carbamazepine, oxcarbazepine, phenobarbital, phenytoin
- rifampicin, rifapentine
- St. John’s wort (Hypericum perforatum)
- mitotane
- enzalutamide
- lumacaftor
4.4. Special warnings and precautions for use
NNRTI substitutions and use of doravirine
Doravirine has not been evaluated in patients with previous virologic failure to any other antiretroviral therapy. NNRTI-associated mutations detected at screening were part of exclusion criteria in the Phase 2b/3-studies. A breakpoint for a reduction in susceptibility, yielded by various NNRTI substitutions, that is associated with a reduction in clinical efficacy has not been established (see section 5.1). There is not sufficient clinical evidence to support the use of doravirine in patients infected with HIV-1 with evidence of resistance to the NNRTI class.
Severe acute exacerbation of hepatitis B in patients co-infected with HIV-1 and HBV
All patients with HIV-1 should be tested for the presence of hepatitis B virus (HBV) before initiating antiretroviral therapy.
Severe acute exacerbations of hepatitis B (e.g., liver decompensated and liver failure) have been reported in patients who are co-infected with HIV-1 and HBV, and have discontinued lamivudine or tenofovir disoproxil, two of the components of Delstrigo. Patients who are co-infected with HIV-1 and HBV should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment with Delstrigo. If appropriate, initiation of anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since post-treatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure.
New onset or worsening renal impairment
Renal impairment, including cases of acute renal failure and Fanconi syndrome (renal tubular injury with severe hypophosphataemia), has been reported with the use of tenofovir disoproxil, a component of Delstrigo.
Delstrigo should be avoided with concurrent or recent use of nephrotoxic medicinal products (e.g., high-dose or multiple nonsteroidal anti-inflammatory medicinal products [NSAIDs]) (see section 4.5). Cases of acute renal failure after initiation of high-dose or multiple NSAIDs have been reported in HIV-infected patients with risk factors for renal dysfunction who appeared stable on tenofovir disoproxil. Some patients required hospitalisation and renal replacement therapy. Alternatives to NSAIDs should be considered, if needed, in patients at risk for renal dysfunction.
Persistent or worsening bone pain, pain in extremities, fractures, and/or muscular pain or weakness may be manifestations of proximal renal tubulopathy and should prompt an evaluation of renal function in at risk patients.
It is recommended that estimated CrCl be assessed in all patients prior to initiating therapy and as clinically appropriate during therapy with Delstrigo. In patients at risk of renal dysfunction, including patients who have previously experienced renal events while receiving adefovir dipivoxil, it is recommended that estimated CrCl, serum phosphorus, urine glucose, and urine protein be assessed prior to initiation of Delstrigo and more frequent renal function monitoring should be assessed as appropriate per the patient’s medical condition during Delstrigo therapy.
Lamivudine and tenofovir disoproxil are primarily excreted by the kidney. Delstrigo should be discontinued if estimated CrCl declines below 50 mL/min as dose interval adjustment required for lamivudine and tenofovir disoproxil cannot be achieved with the fixed dose combination tablet (see section 4.2).
Bone loss and mineralisation defects
Bone mineral density
In clinical trials in HIV-1 infected adults, tenofovir disoproxil was associated with slightly greater decreases in bone mineral density (BMD) and increases in biochemical markers of bone metabolism, suggesting increased bone turnover relative to comparators. Serum parathyroid hormone levels and 1,25 Vitamin D levels were also higher in subjects receiving tenofovir disoproxil. In other studies (prospective and cross-sectional), the most pronounced decreases in BMD were seen in patients treated with tenofovir disoproxil as part of a regimen containing a boosted protease inhibitor.
Bone abnormalities (infrequently contributing to fractures) may be associated with proximal renal tubulopathy.
The effects of tenofovir disoproxil associated changes in BMD and biochemical markers on long-term bone health and future fracture risk are unknown. Assessment of BMD should be considered for HIV-1 infected adult patients who have a history of pathologic bone fracture or other risk factors for osteoporosis or bone loss. Although the effect of supplementation with calcium and Vitamin D was not studied, such supplementation may be beneficial in all patients. If bone abnormalities are suspected, then appropriate consultation should be obtained.
Mineralisation defects
Cases of osteomalacia associated with proximal renal tubulopathy, manifested as bone pain or pain in extremities and which may contribute to fractures, have been reported in association with the use of tenofovir disoproxil . Arthralgias and muscle pain or weakness have also been reported in cases of proximal renal tubulopathy. Hypophosphataemia and osteomalacia secondary to proximal renal tubulopathy should be considered in patients at risk of renal dysfunction who present with persistent or worsening bone or muscle symptoms while receiving products containing tenofovir disoproxil (see section 4.4).
Co-administration with other antiviral products
Doravirine/lamivudine/tenofovir disoproxil must not be co-administered with other medicinal products containing lamivudine, or with medicinal products containing tenofovir disoproxil, or tenofovir alafenamide, or with adefovir dipivoxil (see section 4.5). Doravirine/lamivudine/tenofovir disoproxil should not be administered with doravirine unless needed for dose adjustment (e.g., with rifabutin) (see sections 4.2 and 4.5).
Use with CYP3A inducers
Caution should be given to prescribing doravirine with medicinal products that may reduce the exposure of doravirine (see sections 4.3 and 4.5).
Immune reactivation syndrome
Immune reactivation syndrome has been reported in patients treated with combination antiretroviral therapy. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, autoimmune hepatitis, polymyositis, and GuillainBarré syndrome) have also been reported to occur in the setting of immune reactivation; however, the time to onset is more variable and can occur many months after initiation of treatment.
Lactose
Delstrigo contains lactose monohydrate. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take this medicinal product.
4.5. Interaction with other medicinal products and other forms of interaction
Delstrigo is a complete regimen for the treatment of HIV-1 infection; therefore, Delstrigo should not be administered with other antiretroviral medicinal products. Information regarding potential medicinal product interactions with other antiretroviral medicines is not provided.
Interaction studies have only been performed in adults.
Delstrigo contains doravirine, lamivudine, and tenofovir disoproxil, therefore any interactions identified for these individually are relevant to Delstrigo and are presented in Table 1.
Effects of other medicinal products on doravirine, lamivudine, and tenofovir disoproxil
Doravirine
Doravirine is primarily metabolised by CYP3A, and medicinal products that induce or inhibit CYP3A are expected to affect the clearance of doravirine (see section 5.2). Doravirine/lamivudine/tenofovir disoproxil should not be co-administered with medicinal products that are strong CYP3A enzyme inducers as significant decreases in doravirine plasma concentrations are expected to occur, which may decrease the effectiveness of doravirine/lamivudine/tenofovir disoproxil (see sections 4.3 and 5.2).
Co-administration with the moderate CYP3A inducer rifabutin decreased doravirine concentrations (see Table 1). When Delstrigo is co-administered with rifabutin, a 100 mg dose of doravirine should be given daily, approximately 12 hours after doravirine/lamivudine/tenofovir disoproxil dose (see section 4.2).
Co-administration of doravirine/lamivudine/tenofovir disoproxil with other moderate CYP3A inducers has not been evaluated, but decreased doravirine concentrations are expected. If co-administration with other moderate CYP3A inducers (e.g., debrafenib, lesinurad, bosentan, thioridazine, nafcillin, modafinil, telotristat ethyl) cannot be avoided, a 100 mg dose of doravirine should be administered daily, approximately 12 hours after the administration of doravirine/lamivudine/tenofovir disoproxil dose (see section 4.2).
Co-administration of doravirine/lamivudine/tenofovir disproxil and medicinal products that are inhibitors of CYP3A may result in increased plasma concentrations of doravirine. However, no dose adjustment is needed when doravirine is co-administered with CYP3A inhibitors.
Lamivudine
Because lamivudine is primarily eliminated by the kidneys through a combination of glomerular filtration and active tubular secretion (see section 5.2), co-administration of doravirine/lamivudine/tenofovir disoproxil with medicinal products that reduce renal function or compete for active tubular secretion may increase serum concentrations of lamivudine.
Tenofovir disoproxil
Because tenofovir is primarily eliminated by the kidneys through a combination of glomerular filtration and active tubular secretion (see section 5.2), co-administration of doravirine/lamivudine/tenofovir disoproxil with medicinal products that reduce renal function or compete for active tubular secretion via OAT1, OAT3 or MRP4 may increase serum concentrations of tenofovir.
Due to the tenofovir disoproxil component of doravirine/lamivudine/tenofovir disoproxil, use of the product should be avoided with concurrent or recent use of nephrotoxic medicinal products. Some examples include, but are not limited to, acyclovir, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs (see section 4.4).
Effects of doravirine, lamivudine, and tenofovir disoproxil on other medicinal products
Doravirine
Doravirine at a dose of 100 mg once daily is not likely to have a clinically relevant effect on the plasma concentrations of medicinal products that are dependent on transport proteins for absorption and/or elimination or that are metabolised by CYP enzymes.
However, co-administration of doravirine and the sensitive CYP3A substrate midazolam resulted in a 18% decrease in midazolam exposure, suggesting that doravirine may be a weak CYP3A inducer. Therefore, caution should be used when co-administering doravirine with medicinal products that are sensitive CYP3A substrates that also have a narrow therapeutic window (e.g., tacrolimus and sirolimus).
Lamivudine
Lamivudine does not inhibit or induce CYP enzymes.
Tenofovir
Based on the results of in vitro experiments and the known elimination pathway of tenofovir, the potential for CYP-mediated interactions involving tenofovir with other medicinal products is low.
Interaction table
Table 1 shows the established and other potential medicinal product interactions with the individual components of Delstrigo but is not all inclusive (increase is indicated as ↑, decrease is indicated as ↓, and no change as ↔). For potential medicine product interactions with tenofovir disoproxil or lamivudine, (see sections 4.4 and 5.2).
Table 1. Interactions between the individual components of Delstrigo and other medicinal products:
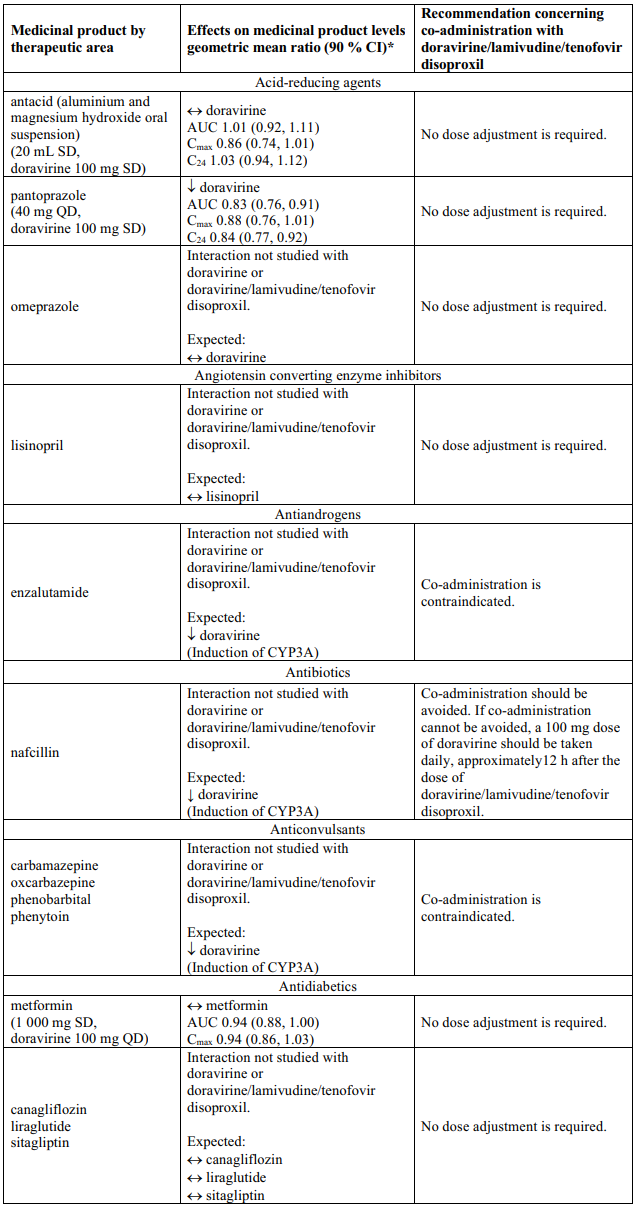
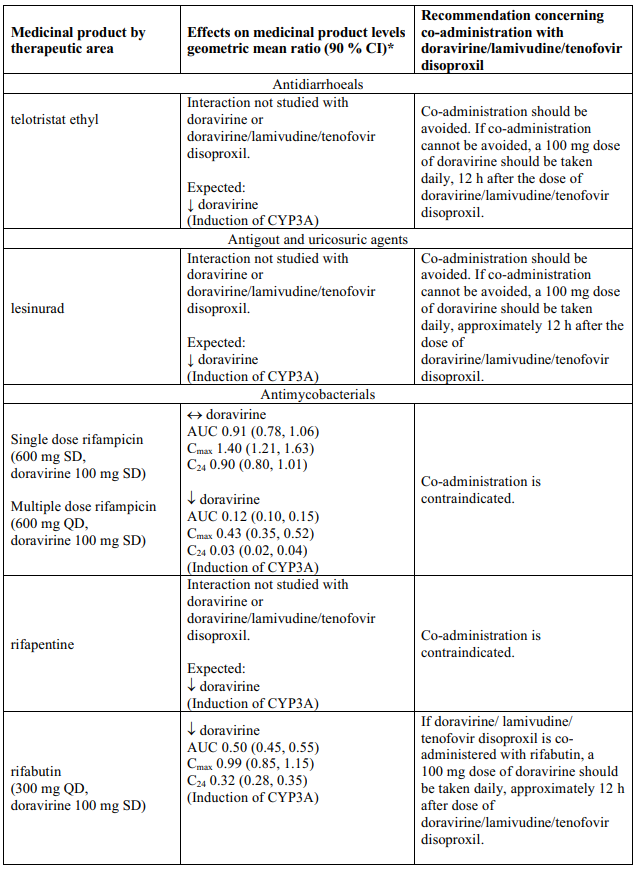
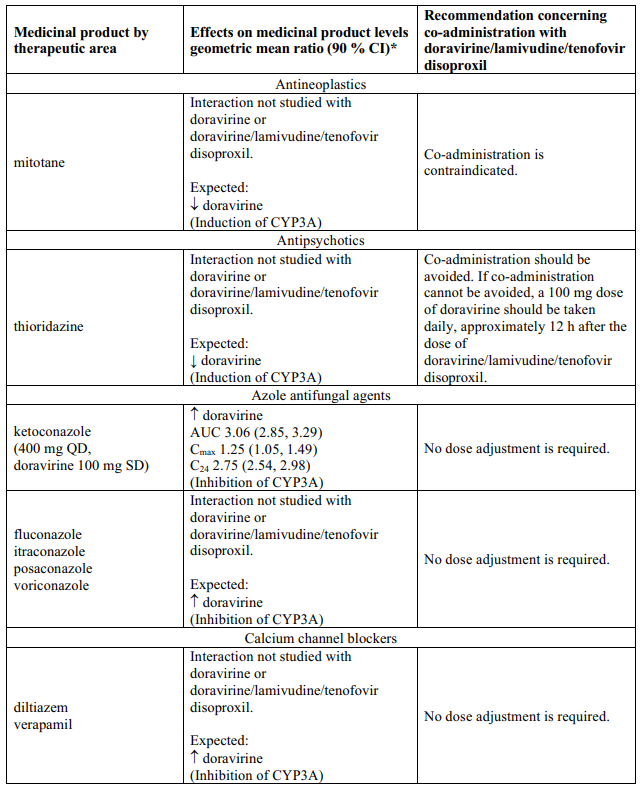

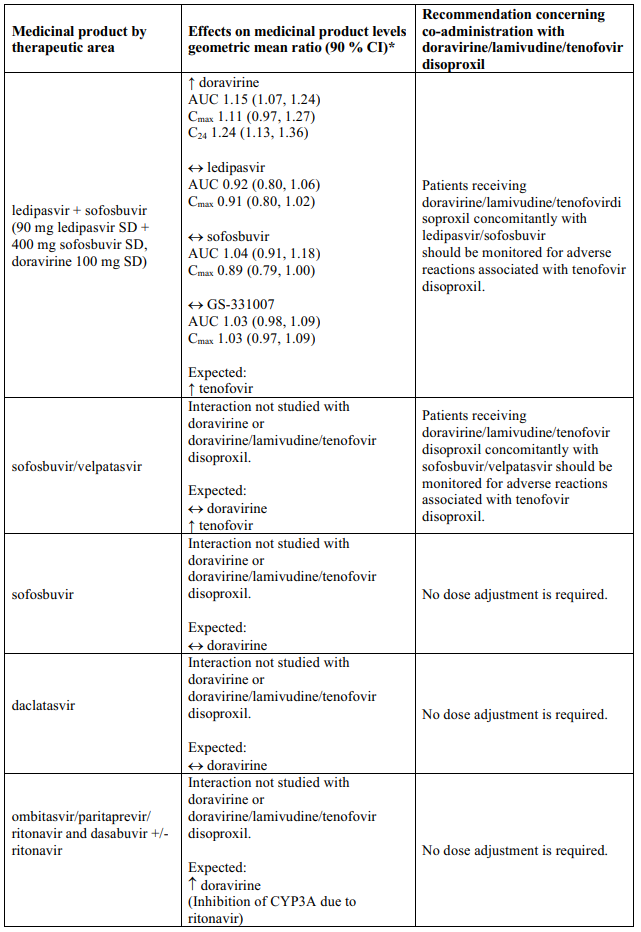
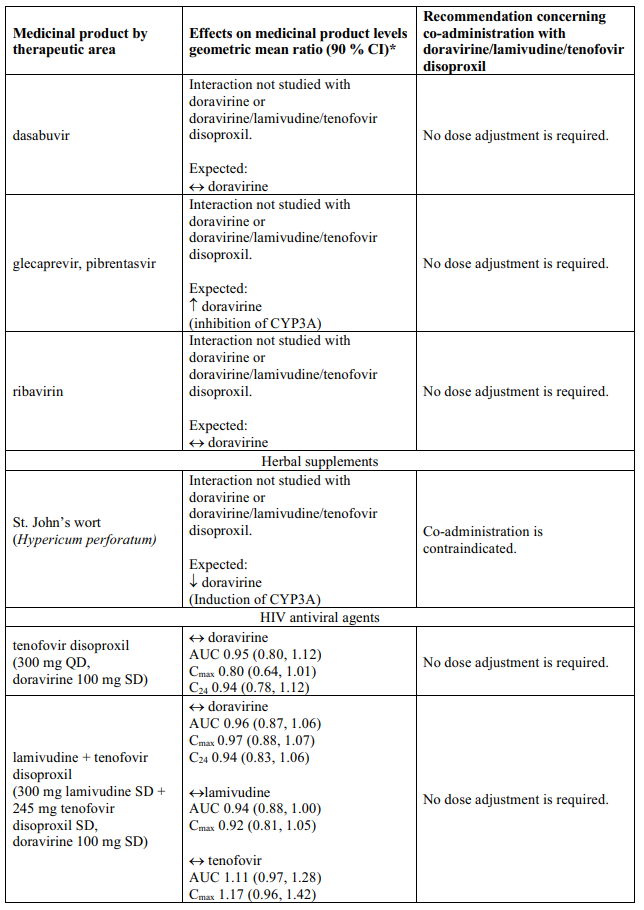
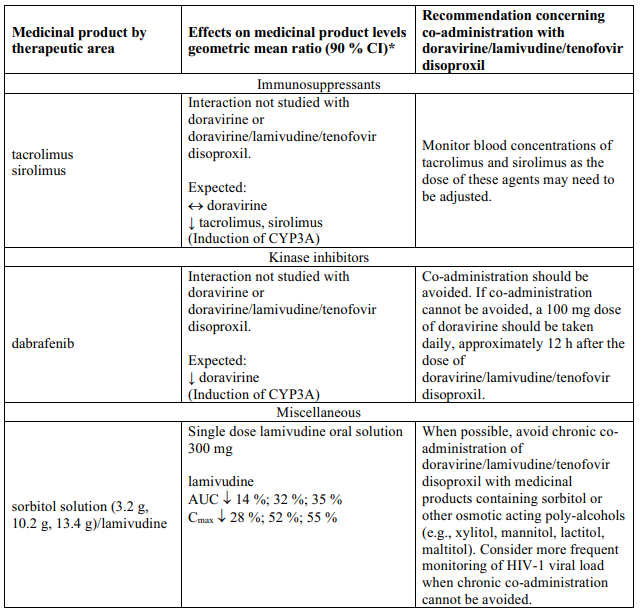
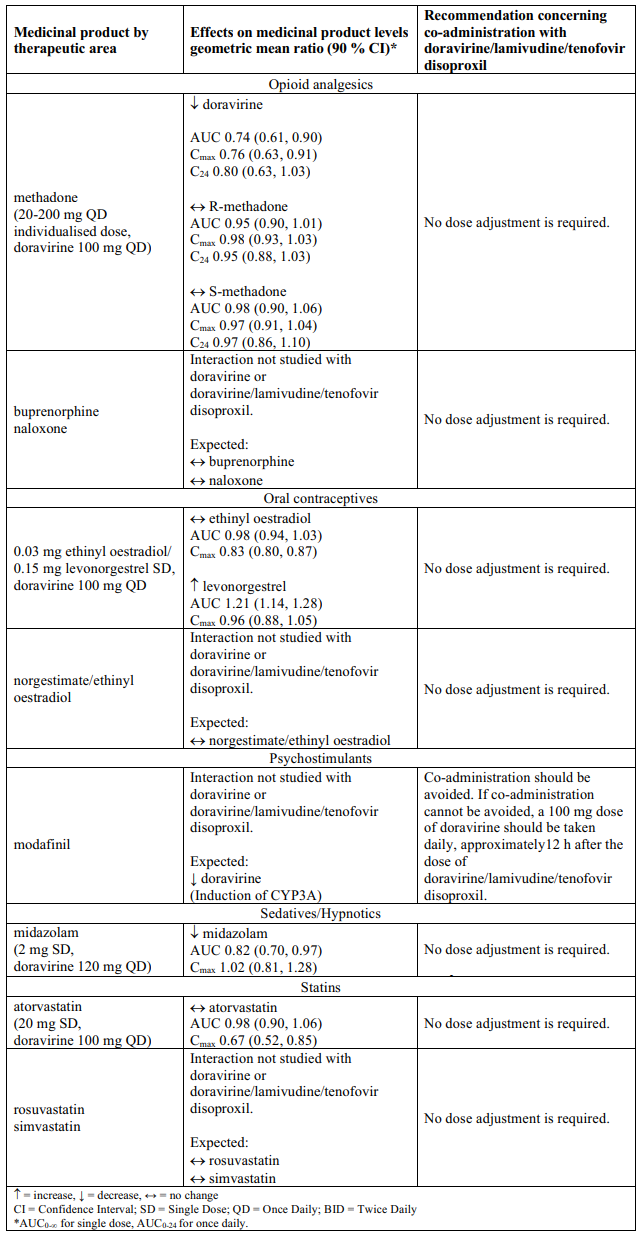
4.6. Fertility, pregnancy and lactation
Pregnancy
There are no or limited amount of data from the use of doravirine in pregnant women. A large amount of data on pregnant women (more than 3 000 outcomes from first trimester) taking the individual active component lamivudine in combination with other antiretrovirals indicates no malformative toxicity. A moderate amount of data on pregnant women (between 300-1 000 pregnancy outcomes) indicate no malformations or foetal/neonatal toxicity associated with tenofovir disoproxil.
Antiretroviral pregnancy registry: To monitor maternal-foetal outcomes in patients exposed to antiretroviral medicinal products while pregnant, an Antiretroviral Pregnancy Registry has been established. Physicians are encouraged to register patients in this registry.
Animal studies with doravirine do not indicate direct or indirect harmful effects with respect to reproductive toxicity (see section 5.3).
Animal studies with tenofovir disoproxil do not indicate direct or indirect harmful effects of tenofovir disoproxil with respect to reproductive toxicity (see section 5.3).
Animal studies with lamivudine showed an increase in early embryonic deaths in rabbits but not in rats (see section 5.3). Placental transfer of lamivudine has been shown to occur in humans. Lamivudine may inhibit cellular DNA replication (see section 5.3). The clinical relevance of this finding is unknown.
As a precautionary measure, it is preferable to avoid the use of Delstrigo during pregnancy.
Breast-feeding
It is unknown whether doravirine is excreted in human milk. Available pharmacodynamic/toxicological data in animals have shown excretion of doravirine in milk (see section 5.3).
Lamivudine has been identified in breast-fed newborns/infants of treated women. Based on more than 200 mother/child pairs treated for HIV, serum concentrations of lamivudine in breast-fed infants of mothers treated for HIV are very low (<4% of maternal serum concentrations) and progressively decrease to undetectable levels when breast-fed infants reach 24 weeks of age. There are no data available on the safety of lamivudine when administered to babies less than three months old.
Tenofovir is excreted in human milk. There is insufficient information on the effects of tenofovir in newborns/infants.
Because of the potential for HIV-1 transmission and the potential for serious adverse reactions in breast-feeding infants, mothers should be instructed not to breast-feed if they are receiving Delstrigo.
Fertility
No human data on the effect of Delstrigo on fertility are available. Animal studies do not indicate harmful effects of doravirine, lamivudine, or tenofovir disoproxil on fertility at exposure levels higher than the exposure in humans at the recommended clinical dose (see section 5.3).
4.7. Effects on ability to drive and use machines
Delstrigo may have a minor influence on the ability to drive and use machines. Patients should be informed that fatigue, dizziness, and somnolence have been reported during treatment with Delstrigo (see section 4.8). This should be considered when assessing a patient’s ability to drive or operate machinery.
4.8. Undesirable effects
Summary of the safety profile
In phase 3 clinical trials with doravirine plus 2 nucleoside analogue reverse transcriptase inhibitors (NRTIs), the most frequently reported adverse reactions were nausea (4%) and headache (3%).
Tabulated summary of adverse reactions
The adverse reactions with doravirine plus 2 NRTIs from Phase 3 clinical trials (DRIVE FORWARD, DRIVE SHIFT and DRIVE AHEAD) are listed below by body system organ class and frequency. Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. Frequencies are defined as very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), or very rare (<1/10,000).
Table 2. Tabulated summary of adverse reactions associated with doravirine/lamivudine/tenofovir disoproxil:
| Frequency | Adverse reactions |
|---|---|
| Infections and infestations | |
| Rare | rash pustular |
| Blood and lymphatic systems disorders | |
| Uncommon | neutropenia*, anaemia*, thrombocytopenia* |
| Very rare | pure red cell aplasia* |
| Metabolism and nutrition disorders | |
| Uncommon | hypophosphataemia, hypokalaemia* |
| Rare | hypomagnesaemia, lactic acidosis* |
| Psychiatric disorders | |
| Common | abnormal dreams, insomnia1 |
| Uncommon | nightmare, depression2, anxiety3, irritability, confusional state, suicidal ideation |
| Rare | aggression, hallucination, adjustment disorder, mood altered, somnambulism |
| Nervous system disorders | |
| Common | headache, dizziness, somnolence |
| Uncommon | disturbance in attention, memory impairment, paraesthesia, hypertonia, poor quality sleep |
| Very rare | peripheral neuropathy* (or paraesthesia) |
| Vascular disorders | |
| Uncommon | hypertension |
| Respiratory, thoracic and mediastinal disorders | |
| Common | cough*, nasal symptoms* |
| Rare | dyspnoea, tonsillar hypertrophy |
| Gastrointestinal disorders | |
| Common | nausea, diarrhoea, abdominal pain4, vomiting, flatulence |
| Uncommon | constipation, abdominal discomfort5, abdominal distension, dyspepsia, faeces soft6, gastrointestinal motility disorder7, pancreatitis* |
| Rare | rectal tenesmus |
| Hepatobiliary disorders | |
| Rare | hepatic steatosis*, hepatitis* |
| Skin and subcutaneous tissue disorders | |
| Common | alopecia*, rash8 |
| Uncommon | pruritus |
| Rare | dermatitis allergic, rosacea, angioedema* |
| Musculoskeletal and connective tissue disorders | |
| Common | muscle disorders* |
| Uncommon | myalgia, arthralgia, rhabdomyolysis*†, muscular weakness*† |
| Rare | musculoskeletal pain, osteomalacia* (manifested as bone pain and infrequently contributing to fractures), myopathy* |
| Renal and urinary disorders | |
| Uncommon | increased creatinine*, proximal renal tubulopathy (including Fanconi syndrome)* |
| Rare | acute kidney injury, renal disorder, calculus urinary, nephrolithiasis, acute renal failure*, renal failure*, acute tubular necrosis*, nephritis* (including acute interstitial), nephrogenic diabetes insipidus* |
| General disorders and administration site conditions | |
| Common | fatigue, fever* |
| Uncommon | asthenia, malaise |
| Rare | chest pain, chills, pain, thirst |
| Investigations | |
| Common | alanine aminotransferase increased9 |
| Uncommon | aspartate aminotransferase increased, lipase increased, amylase increased, haemoglobin decreased |
| Rare | blood creatine phosphokinase increased |
* This adverse reaction was not identified as an adverse reaction associated with doravirine from the Phase 3 clinical studies (DRIVE-FORWARD, DRIVE-AHEAD, DRIVE-SHIFT), but is included in this table as an adverse reaction based on the Summary of Product Characteristics of 3TC and/or TDF. The highest frequency category reported in the 3TC or TDF Summary of Product Characteristics is used.
† This adverse reaction may occur as a consequence of proximal renal tubulopathy. It is not considered to be causally
associated with tenofovir disoproxil in the absence of this condition.
1 insomnia includes: insomnia, initial insomnia and sleep disorder.
2 depression includes: depression, depressed mood, major depression, and persistent depressive disorder.
3 anxiety includes: anxiety and generalised anxiety disorder.
4 abdominal pain includes: abdominal pain, and abdominal pain upper.
5 abdominal discomfort includes: abdominal discomfort, and epigastric discomfort.
6 faeces soft includes: faeces soft and abnormal faeces.
7 gastrointestinal motility disorder includes: gastrointestinal motility disorder, and frequent bowel movements.
8 rash includes: rash, rash macular, rash erythematous, rash generalised, rash maculo-papular, rash papular, and urticarial.
9 alanine aminotransferase increased includes: alanine aminotransferase increased andhepatocellular injury.
Immune reactivation syndrome
In HIV-infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
Lactic acidosis
Cases of lactic acidosis have been reported with tenofovir disoproxil alone or in combination with other antiretrovirals. Patients with predisposing factors such as patients with decompensated liver disease, or patients receiving concomitant medicinal products known to induce lactic acidosis are at increased risk of experiencing severe lactic acidosis during tenofovir disoproxil treatment, including fatal outcomes.
Paediatric population
The safety of doravirine/lamivudine/tenofovir disoproxil was evaluated in 45 HIV-1 infected virologically suppressed or treatment-naïve paediatric patients 12 to less than 18 years of age through Week 48 in an open-label trial (IMPAACT 2014 (Protocol 027)). The safety profile in paediatric subjects was similar to that in adults.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
6.2. Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.