FRUZAQLA Hard capsule Ref.[111509] Active ingredients: Fruquintinib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Takeda Pharmaceuticals International AG Ireland Branch, Block 2 Miesian Plaza, 50-58 Baggot Street Lower, Dublin 2, D02 HW68, Ireland, medinfoEMEA@takeda.com
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, vascular endothelial growth factor receptor (VEGFR)-tyrosine kinase inhibitors
ATC code: L01EK04
Mechanism of action and pharmacodynamic effects
Fruquintinib is a selective tyrosine kinase inhibitor of VEGFR-1, -2, and -3 with antitumor effects resulting from suppression of tumour angiogenesis.
Cardiac electrophysiology
No prolongation of heart rate-corrected QT (QTc) interval (>10 milliseconds) was observed at the recommended dosage of fruquintinib. A concentration-QT analysis (N=205) showed no evidence of an association between fruquintinib plasma concentrations and changes in QTc interval from baseline.
Clinical efficacy and safety
The efficacy and safety of fruquintinib plus best supportive care (BSC) was evaluated in a randomised, placebo-controlled, double-blind, phase III study (FRESCO-2) in patients with mCRC previously 13 treated with but not limited to oxaliplatin or irinotecan-based chemotherapies. The clinical efficacy of fruquintinib in the FRESCO-2 study is described below.
FRESCO-2 Study
The clinical efficacy and safety of fruquintinib were evaluated in a global, randomised, double-blind, placebo-controlled, multicentre, phase III study (FRESCO-2) in 691 patients with mCRC who had been previously treated with standard approved therapies including fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy; an anti-VEGF biological therapy; an anti-EGFR therapy if RAS wild-type, and have progressed on or had intolerance to trifluridine/tipiracil and/or regorafenib. Patients were considered intolerant to trifluridine/tipiracil or regorafenib if they received at least 1 dose of either agent and were discontinued from therapy for reasons other than progressive disease. Patients with MSI-H or dMMR tumours were previously treated with immune checkpoint inhibitors, and patients with BRAF V600E mutant tumours were previously treated with a BRAF inhibitor, if approved and available in the patients' respective country or region. Randomisation was stratified by prior therapy (trifluridine/tipiracil vs. regorafenib vs. both trifluridine/tipiracil and regorafenib), RAS status (wild-type vs. mutant), and duration of metastatic disease (≤18 months vs. >18 months).
Patients with an Eastern Cooperative Oncology Group (ECOG) performance status ≥2, left ventricular fraction ≤50%, systolic blood pressure >140 mm Hg or diastolic blood pressure >90 mm Hg, urine protein ≥1 g/24h, or body weight <40 kg were excluded. The primary efficacy endpoint was overall survival (OS). The key secondary efficacy endpoint was progression-free survival (PFS; as assessed by the investigator using Response Evaluation Criteria in Solid Tumours [RECIST], version 1.1) and other supportive secondary endpoints included disease control rate.
In total, 691 patients were randomised (2:1) to receive fruquintinib 5 mg orally once daily (N=461) plus BSC or placebo orally once daily (N=230) plus BSC (hereafter denoted as fruquintinib and placebo, respectively), for 21 days on therapy followed by 7 days off-therapy in a 28-day treatment cycle.
Among the 691 randomised patients, the median age was 64 years (range: 25 to 86), with 47% ≥65 years of age. 55.7% of patients were male, 80.9% were White, and had an ECOG performance status of 0 (43.1%) or 1 (56.9%). Tumour RAS wild-type was reported in 36.9% of patients at study entry. The median duration of metastatic disease of 39 months (range: 6 months to 16.1 years). The median number of prior lines of therapy for metastatic disease was 4 (range: 2 to 16). In addition to treatment with fluoropyrimidine, oxaliplatin, and irinotecan-based chemotherapy, 96.4% of patients received prior anti-VEGF therapy, 38.8% received prior anti-EGFR therapy, 52.2% received trifluridine/tipiracil, and 8.4% received regorafenib, and 39.4% received both trifluridine/tipiracil and regorafenib, 4.6% received immunotherapy, and 2.3% received BRAF inhibitor.
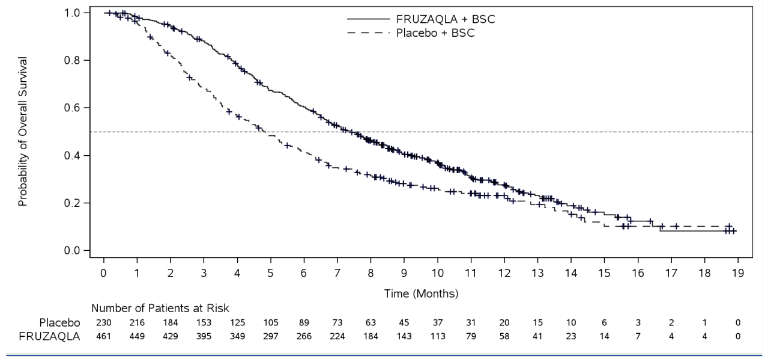
In FRESCO-2, the addition of fruquintinib to BSC resulted in a statistically significant improvement in OS and PFS compared to placebo plus BSC (see Table 4 and Figure 1).
Table 4. Efficacy results from the FRESCO-2 study:
| Endpoint | Fruquintinib (N=461) | Placebo (N=230) |
|---|---|---|
| OS | ||
| Median in months (95% CI) | 7.4 (6.7, 8.2) | 4.8 (4.0, 5.8) |
| Hazard Ratio1 (95% CI) | 0.66 (0.55, 0.80) | |
| p-value2 | <0.001 | |
| PFS3 | ||
| Median in months (95% CI) | 3.7 (3.5, 3.8) | 1.8 (1.8, 1.9) |
| Hazard Ratio1 (95% CI) | 0.32 (0.27 to 0.39) | |
| p-value2 | <0.001 | |
Abbreviations: CI=confidence interval; HR=hazard ratio; N=number of patients; OS=overall survival; PFS=progression-free survival
The median OS and PFS were calculated using the Kaplan-Meier method.
1 The HR and its 95% CI were estimated using stratified Cox’s proportional hazards model (accounting for the stratification factors), in which the treatment arm is the only covariate in the model.
2 p-value (2-sided) was calculated using the stratified log-rank test to account for the stratification factors.
3 Assessed by the investigator using RECIST, version 1.1.
Figure 1. Kaplan-Meier curve for overall survival in FRESCO-2 study:
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with FRUZAQLA in all subsets of the paediatric population in metastatic colorectal cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
After oral administration of fruquintinib, the median time to achieve peak plasma fruquintinib concentration (Tmax) was approximately 2 hours. Fruquintinib showed a second absorption peak approximately 24 hours after drug administration. Following repeat once-daily dosing, fruquintinib exposure (Cmax and AUC0-24h) increased in a dose-proportional manner across the dose range of 1 to 6 mg (0.2 to 1.2 times the recommended dosage). Following administration of fruquintinib 5 mg once daily for 21 days with 7 days off of each 28-day cycle in patients with advanced solid tumours, steady state of fruquintinib was achieved after 14 days, and the mean accumulation based on AUC0-24h was 4-fold relative to a single dose. At the recommended dose of 5 mg of fruquintinib, the geometric mean (CV) Cmax and AUC0-24h for fruquintinib at steady state were 300 ng/mL (28) and 5880 ng*h/mL (29%), respectively.
Effect of food
Compared to the fasting state, a high-fat meal had no clinically meaningful effect on fruquintinib pharmacokinetics in healthy subjects. Fruquintinib can be administered with or without food.
Distribution
The apparent volume of distribution of fruquintinib is approximately 48.5 L. Plasma protein binding of fruquintinib is approximately 95% in vitro and mainly bound to human serum albumin.
Biotransformation
Fruquintinib is metabolised by multiple enzymes, including CYP450 (CYP3A and CYP2C subfamilies) and non-CYP450 enzyme systems. The in vivo metabolism and mass balance study of [14C] labelled fruquintinib showed that fruquintinib mainly exists in human plasma in its unchanged form, accounting for approximately 72% of total exposure in the plasma, and the CYP3A4-mediated N-demethyl metabolite of fruquintinib account for approximately 17% of total exposure in plasma. Other metabolic pathways include multi-site mono-oxidation, O-demethylation, N-demethylation, O-dequinazoline ring, and amide hydrolysis. The phase II metabolites are mainly glucuronic acid and sulphuric acid conjugates of phase I products.
In vitro studies
Cytochrome P450 enzymes:
CYP3A4 was the main enzyme among the CYP isoforms involved in the metabolism of fruquintinib, with minor contributions from CYP2C8, CYP2C9 and CYP2C19. Fruquintinib is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A, or an inducer of CYP1A2, CYP2B6, CYP3A at therapeutically relevant concentrations.
Transporter systems:
Fruquintinib is not a substrate of P-glycoprotein (P-gp), organic anion transport protein (OATP)1B1, or OATP1B3. Fruquintinib inhibited P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) in a dose-dependent manner in vitro and demonstrated pH-dependent aqueous solubility. Fruquintinib is not an inhibitor of OATP1B1, OATP1B3, organic anion transporter (OAT)1, OAT3, organic cation transporter (OCT)2, multidrug and toxin extrusion protein (MATE)1, or MATE2-K at therapeutically relevant concentrations.
Elimination
The apparent clearance (CL/F) of fruquintinib is 14.8 mL/min at steady-state after once daily dosing in patients with advanced solid tumours. The mean elimination half-life of fruquintinib is approximately 42 hours.
Following administration of a single 5 mg radiolabelled fruquintinib in healthy subjects, approximately 60% of the dose was recovered in urine (0.5% of the dose as unchanged fruquintinib), and 30% of the dose was recovered in faeces (5% of the dose as unchanged fruquintinib).
Special populations
Renal impairment
Based on the population pharmacokinetic analyses, mild to moderate renal impairment (creatinine clearance [CrCL] 30 to 89 mL/min) had no clinically meaningful impact on fruquintinib pharmacokinetics. In a pharmacokinetic study, unbound fruquintinib AUC0-inf and Cmax were similar in subjects with moderate (CrCL 30–59 mL/min, N=8) or severe (CrCL 15–29 mL/min, N=8) renal impairment as compared to subjects with normal renal function (CrCL ≥90 mL/min, N=8).
Hepatic impairment
No clinically meaningful differences in the pharmacokinetics of fruquintinib were observed between patients with normal hepatic function and patients with mild (total bilirubin ≤ ULN with AST greater than ULN or total bilirubin >1 to 1.5 times ULN with any AST) hepatic impairment based on population pharmacokinetic analyses. Based on a dedicated hepatic impairment pharmacokinetic study, following administration of a single 2 mg oral dose of fruquintinib, no clinically meaningful differences in the dose-normalised AUC of fruquintinib were observed in subjects with moderate (Child Pugh B) hepatic impairment compared to subjects with normal hepatic function.
Age, body weight, gender or race
Population pharmacokinetic analyses showed that age (18 to 82 years), body weight (48 to 108 kg), gender or race had no clinically relevant impact on the pharmacokinetics of fruquintinib.
Paediatric population
No pharmacokinetic studies were performed with fruquintinib in patients under 18 years of age.
5.3. Preclinical safety data
In repeat dose and reproductive toxicity studies, toxicity was observed at fruquintinib average plasma concentrations below the expected human therapeutic concentrations.
Repeat dose toxicity
In repeat dose animal toxicity studies, the main target organ effects were identified in the gastrointestinal tract, hepatobiliary system, immune system, skeletal system (femur and teeth), kidneys, hematopoietic system, and adrenal gland and appear related to the pharmacology of VEGFR inhibition and/or disruption of VEGF signalling pathway. All findings were reversible after 4 weeks without treatment, apart from the skeletal system (broken/lost teeth).
Impairment of fertility
In a fertility and early embryonic development study in rats, male and female reproductive indices were decreased at exposures approximately 3.2 and 0.8-fold the human AUC, respectively. Dose-dependent increases in pre-implantation loss were observed in the same study.
Reproductive toxicity
In an embryo-foetal developmental study in rats, embryotoxic and teratogenic effects were observed at subclinical exposure levels in the absence of excessive maternal toxicity, consisting of foetal external, visceral, and skeletal malformations. Malformations affected primarily the head, tail, tongue, blood vessels, heart, thymus, and developing skeleton (notably vertebrae).
Genotoxicity
No evidence of genotoxicity was observed in in vitro and in vivo studies.
Carcinogenesis
Carcinogenicity studies have not been performed with fruquintinib.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.