NAVERUCLIF Powder for dispersion for infusion Ref.[109403] Active ingredients: Paclitaxel
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Accord Healthcare S.L.U., World Trade Center, Moll de Barcelona, s/n, Edifici Est 6ª planta, 08039 Barcelona, Spain
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, plant alkaloids and other natural products, taxanes,
ATC code: L01CD01
Mechanism of action
Paclitaxel is an antimicrotubule agent that promotes the assembly of microtubules from tubulin dimers and stabilises microtubules by preventing depolymerisation. This stability results in the inhibition of the normal dynamic reorganisation of the microtubule network that is essential for vital interphase and mitotic cellular functions. In addition, paclitaxel induces abnormal arrays or "bundles" of microtubules throughout the cell cycle and multiple asters of microtubules during mitosis.
Naveruclif contains human serum albumin-paclitaxel nanoparticles of approximately 180 nm in size, where the paclitaxel is present in a non-crystalline, amorphous state. Upon intravenous administration, the nanoparticles dissociate rapidly into soluble, albumin bound paclitaxel complexes of approximately 10 nm in size. Albumin is known to mediate endothelial caveolar transcytosis of plasma constituents, and in vitro studies demonstrated that the presence of albumin enhances transport of paclitaxel across endothelial cells. It is hypothesised that this enhanced transendothelial caveolar transport is mediated by the gp-60 albumin receptor, and that there is enhanced accumulation of paclitaxel in the area of tumour due to the albumin-binding protein Secreted Protein Acidic Rich in Cysteine (SPARC).
Clinical efficacy and safety
Breast cancer
Data from 106 patients accrued in two single-arm open-label studies and from 454 patients treated in a randomised Phase III comparative study are available to support the use of human serum albuminpaclitaxel nanoparticles in metastatic breast cancer. This information is presented below.
Single-arm open-labelstudies
In one study, human serum albumin-paclitaxel nanoparticles were administered as a 30-minute infusion at a dose of 175 mg/m² to 43 patients with metastatic breast cancer. The second trial utilised a dose of 300 mg/m² as a 30-minute infusion in 63 patients with metastatic breast cancer. Patients were treated without steroid pre-treatment or planned G-CSF support. Cycles were administered at 3-week intervals. The response rates in all patients were 39.5% (95% CI: 24.9%-54.2%) and 47.6% (95% CI: 35.3%-60.0%), respectively. The median time to disease progression was 5.3 months (175 mg/m²; 95% CI: 4.6-6.2 months) and 6.1 months (300 mg/m²; 95% CI: 4.2-9.8 months).
Randomised comparative study
This multi-centre trial was conducted in patients with metastatic breast cancer, who were treated every 3 weeks with single-agent paclitaxel, either as solvent-based paclitaxel 175 mg/m² given as a 3-hour infusion with premedication to prevent hypersensitivity (N = 225), or as human serum albumin-paclitaxel nanoparticles 260 mg/m² given as a 30 minute infusion without premedication (N = 229).
Sixty-four percent of patients had impaired performance status (ECOG 1 or 2) at study entry; 79% had visceral metastases; and 76% had > 3 sites of metastases. Fourteen percent of the patients had not received prior chemotherapy; 27% had received chemotherapy in the adjuvant setting only, 40% in the metastatic setting only, and 19% in both metastatic and adjuvant settings. Fifty-nine percent received study medicinal product as second or greater than second-line therapy. Seventyseven percent of the patients had been previously exposed to anthracyclines.
Results for overall response rate and time to disease progression, and progression-free survival and survival for patients receiving > 1st-line therapy, are shown below.
Table 8. Results for overall response rate, median time to disease progression, and progressionfree survival as assessed by the investigator:
| Efficacy variable | Human serum albumin-paclitaxel nanoparticles (260 mg/m²) | Solvent-based paclitaxel (175 mg/m²) | p-value |
|---|---|---|---|
| Response rate [95% CI] (%) | |||
| > 1st-line therapy | 26.5 [18.98, 34.05] (n = 132) | 13.2 [7.54, 18.93] (n = 136) | 0.006a |
| *Median time to disease progression [95% CI] (weeks) | |||
| > 1st-line therapy | 20.9 [15.7, 25.9] (n = 131) | 16.1 [15.0, 19.3] (n = 135) | 0.011b |
| *Median progression free survival [95% CI] (weeks) | |||
| > 1st-line therapy | 20.6 [15.6, 25.9] (n = 131) | 16.1 [15.0, 18.3] (n = 135) | 0.010b |
| *Survival [95% CI] (weeks) | |||
| > 1st-line therapy | 56.4 [45.1, 76.9] (n = 131) | 46.7 [39.0, 55.3] (n = 136) | 0.020b |
* This data is based on Clinical Study Report: CA012-0 Addendum dated Final (23 March-2005)
a Chi-squared test
b Log-rank test
Two hundred and twenty nine patients treated with human serum albumin-paclitaxel nanoparticles in the randomized, controlled clinical trial were evaluated for safety. Neurotoxicity to paclitaxel was evaluated through improvement by one grade for patients experiencing Grade 3 peripheral neuropathy at any time during therapy. The natural course of peripheral neuropathy to resolution to baseline due to cumulative toxicity of human serum albumin-paclitaxel nanoparticles after > 6 courses of treatment was not evaluated and remains unknown.
Pancreatic adenocarcinoma
A multicenter, multinational, randomized, open-label study was conducted in 861 patients to compare human serum albumin-paclitaxel nanoparticles/gemcitabine versus gemcitabine monotherapy asfirstline treatment in patients with metastatic adenocarcinoma of the pancreas. Human serum albumin-paclitaxel nanoparticles were administered to patients (N = 431) as an intravenous infusion over 30-40 minutes at a dose of 125 mg/m² followed by gemcitabine as an intravenous infusion over 30- 40 minutes at a dose of 1000 mg/m² given on Days 1, 8 and 15 of each 28-day cycle. In the comparator treatment arm, gemcitabine monotherapy was administered to patients (N = 430) in accordance with the recommended dose and regimen. Treatment was administered until disease progression or development of an unacceptable toxicity. Of the 431 patients with pancreatic adenocarcinoma who were randomized to receive human serum albumin-paclitaxel nanoparticles in combination with gemcitabine, the majority (93%) were white, 4% were black and 2% were Asian. 16% had a Karnofsky Performance Status of 100; 42% had a KPS of 90; 35% had a KPS of 80; 7% had a KPS of 70; and <1% of patients had a KPS of below 70. Patients with high cardiovascular risk, history of peripheral artery disease and/or of connective tissue disorders and/or interstitial lung disease were excluded from the study.
Patients received a median treatment duration of 3.9 months in the human serum albumin-paclitaxel nanoparticles/gemcitabine arm and 2.8 months in the gemcitabine arm. 32% of patients in the human serum albumin-paclitaxel nanoparticles/gemcitabine arm compared with 15% of patients in the gemcitabine arm received 6 or more months of treatment. For the treated population, the median relative dose intensity for gemcitabine was 75% in the human serum albumin-paclitaxel nanoparticles/gemcitabine arm and 85% in the gemcitabine arm. The median relative dose intensity of human serum albumin-paclitaxel nanoparticles was 81%. A higher median cumulative dose of gemcitabine was delivered in the human serum albumin-paclitaxel nanoparticles/gemcitabine arm (11400 mg/m²) when compared with the gemcitabine arm (9000 mg/m²).
The primary efficacy endpoint was overall survival (OS). The key secondary endpoints were progression- free survival (PFS) and overall response rate (ORR), both assessed by independent, central, blinded radiological review using RECIST guidelines (Version 1.0).
Table 9. Efficacy results from randomized study in patients with pancreatic adenocarcinoma (Intent-to-treatpopulation):
| Human serum albumin-paclitaxel nanoparticles (125 mg/m²)/gemcitabine (N=431) | Gemcitabine (N=430) | |
|---|---|---|
| Overall Survival | ||
| Number of deaths (%) | 333 (77) | 359 (83) |
| Median Overall Survival, months (95% CI) | 8.5 (7.89, 9.53) | 6.7 (6.01, 7.23) |
| HRA+G/G (95% CI)a | 0.72 (0.617, 0.835) | |
| P-valueb | <0.0001 | |
| Survival Rate % (95% CI) at | ||
| 1 Year | 35% (29.7, 39.5) | 22% (18.1, 26.7) |
| 2 Year | 9% (6.2, 13.1) | 4% (2.3, 7.2) |
| 75th Percentile Overall Survival (months) | 14.8 | 11.4 |
| Progression-free Survival | ||
| Death or progression, n (%) | 277 (64) | 265 (62) |
| Median Progression-free Survival, months (95% CI) | 5.5 (4.47, 5.95) | 3.7 (3.61, 4.04) |
| HRA+G/G (95% CI)a | 0.69 (0.581, 0.821) | |
| P-valueb | <0.0001 | |
| Overall Response Rate | ||
| Confirmed complete or partial overall response, n (%) | 99 (23) | 31 (7) |
| 95% CI | 19.1, 27.2 | 5.0, 10.1 |
| pA+G/pG (95% CI) | 3.19 (2.178, 4.662) | |
| P-value (chi-square test) | <0.0001 | |
CI = confidence interval, HRA+G/G = hazard ratio of human serum albumin-paclitaxel nanoparticles+gemcitabine/gemcitabine, pA+G/pG=response rate ratio of human serum albuminpaclitaxel nanoparticles+gemcitabine/gemcitabine
a stratified Cox proportional hazard model
b stratified log-rank test, stratified by geographic region (North America versus others), KPS (70 to 80 versus 90 to 100), and presence of liver metastasis (yes versus no).
There was a statistically significant improvement in OS for patients treated with human serum albuminpaclitaxel nanoparticles/gemcitabine versus gemcitabine alone, with 1.8 months increase in median OS, 28% overall reduction in risk of death, 59% improvement in 1-year survival, and 125% improvement in 2-year survival rates.
Figure 1. Kaplan-Meier curve of overall survival (intent-to-treat population):
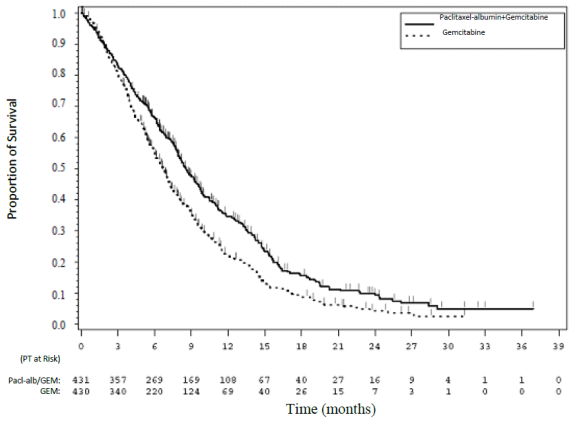
Treatment effects on OS favoured the human serum albumin-paclitaxel nanoparticles/gemcitabine arm across the majority of pre-specified subgroups (including gender, KPS, geographic region, primary location of pancreatic cancer, stage at diagnosis, presence of liver metastases, presence of peritoneal carcinomatosis, prior Whipple procedure, presence of biliary stent at baseline, presence of pulmonary metastases, and number of metastatic sites). For patients ≥ 75 years of age in the human serum albuminpaclitaxel nanoparticles/gemcitabine and gemcitabine arms the survival Hazard Ratio (HR) was 1.08 (95% CI 0.653, 1. 797). For patients with normal baseline CA 19-9 levels the survival HR was 1.07 (95% CI 0.692, 1.661).
There was a statistically significant improvement in PFS for patients treated with human serum albumin-paclitaxel nanoparticles/gemcitabine versus gemcitabine alone, with 1.8 months increase in median PFS.
Non-small cell lung cancer
A multicenter, randomized, open-label study was conducted in 1052 chemotherapy-naive patients with Stage IIIb/IV non-small cell lung cancer. The study compared human serum albumin-paclitaxel nanoparticles in combination with carboplatin versus solvent-based paclitaxel in combination with carboplatin as first-line treatment in patients with advanced non-small cell lung cancer. Over 99% of patients had an ECOG (Eastern Cooperative Oncology Group) performance status of 0 or 1. Patients with pre-existing neuropathy of Grade ≥ 2 or serious medical risk factors involving any of the major organ systems were excluded. Human serum albumin-paclitaxel nanoparticles were administered to patients (N=521) as an intravenous infusion over 30 minutes at a dose of 100 mg/m² on Days 1, 8 and 15 of each 21-day cycle without any steroid premedication and without granulocyte colony stimulating factor prophylaxis. Beginning immediately after the end of human serum albumin-paclitaxel nanoparticles administration, carboplatin at a dose of AUC = 6 mg•min/mL was administered intravenously on Day 1 only of each 21-day cycle. Solvent-based paclitaxel was administered to patients (N=531) at a dose of 200 mg/m² as an intravenous infusion over 3 hours with standard premedication, immediately followed by carboplatin administered intravenously at AUC = 6 mg•min/mL. Each drug was administered on Day 1 of each 21-day cycle. In both study arms treatment was administered until disease progression or development of an unacceptable toxicity. Patients received a median of 6 cycles of treatment in both study arms.
The primary efficacy endpoint was overall response rate defined as the percentage of patients who achieved an objective confirmed complete response or partial response based on an independent, central, blinded radiological review using RECIST (Version 1.0). Patients in the human serum albuminpaclitaxel nanoparticles/carboplatin arm had a significantly higher overall response rate compared with patients in the control arm: 33% versus 25%, p = 0.005 (Table 10). There was a significant difference in overall response rate in the human serum albumin-paclitaxel nanoparticles/carboplatin arm compared to the control arm in patients with non-small cell lung cancer of squamous histology (N=450, 41% vs. 24%, p<0.001), however this difference did not translate into a difference in PFS or OS. There was no difference in ORR between the treatment arms in patients with non-squamous histology (N=602, 26% vs 25%, p=0.808).
Table 10. Overall response rate in randomized non-small cell lung cancer trial (intent-to-treat population):
| Efficacy Parameter | Human serum albumin-paclitaxel nanoparticles (100 mg/m²/week) + carboplatin (N=521) | Solvent-based paclitaxel (200 mg/m² every 3 weeks) + carboplatin (N=531) |
|---|---|---|
| Overall Response Rate (independent review) | ||
| Confirmed complete or partial overall response, n (%) | 170 (33%) | 132 (25%) |
| 95% CI (%) | 28.6, 36.7 | 21.2, 28.5 |
| pA/pT (95.1% CI) | 1.313 (1.082, 1.593) | |
| P-valuea | 0.005 | |
CI = confidence interval; HRA/T = hazard ratio of human serum albumin-paclitaxel nanoparticles/carboplatin to solvent-based paclitaxel/carboplatin;
pA/pT = response rate ratio of human serum albumin-paclitaxel nanoparticles/carboplatin to solventbased paclitaxel/carboplatin.
a P-value is based on a chi-square test.
There was no statistically significant difference in progression-free survival (by blinded radiologist assessment) and overall survival between the two treatment arms. A non-inferiority analysis was conducted for PFS and OS, with a pre-specified non-inferiority margin of 15%. The non-inferiority criterion was met for both PFS and OS with the upper bound of the 95% confidence interval for the associated hazard ratios being less than 1.176 (Table 11).
Table 11. Non-inferiority analyses on progression-free survival and overall survival in randomized non-small cell lung cancer trial (intent-to-treat population):
| Efficacy Parameter | Human serum albuminpaclitaxel nanoparticles (100 mg/m²/week) + carboplatin (N=521) | Solvent-based paclitaxel (200 mg/m² every 3 weeks) + carboplatin (N=531) |
|---|---|---|
| Progression-free Survivala (independent review) | ||
| Death or progression, n (%) | 429 (82%) | 442 (83%) |
| Median PFS (95% CI) (months) | 6.8 (5.7, 7.7) | 6.5 (5.7, 6.9) |
| HRA/T (95% CI) | 0.949 (0.830, 1.086) | |
| Overall Survival | ||
| Number of deaths, n (%) | 360 (69%) | 384 (72%) |
| Median OS (95% CI) (months) | 12.1 (10.8, 12.9) | 11.2 (10.3, 12.6) |
| HRA/T (95.1% CI) | 0.922 (0.797, 1.066) | |
CI = confidence interval; HRA/T = hazard ratio of human serum albumin-paclitaxel nanoparticles/carboplatin to solvent-based paclitaxel/carboplatin;
pA/pT = response rate ratio of human serum albumin-paclitaxel nanoparticles/carboplatin to solventbased paclitaxel/carboplatin.
aPer EMA methodological considerations for PFS endpoint, missing observations or initiation of subsequent new therapy were not used for censoring.
Paediatric population
Safety and effectiveness in paediatric patients have not been established (see section 4.2).
Study ABI-007-PST-001, a Phase 1/2, multicenter, open-label, dose-finding study to assess the safety, tolerability and preliminary efficacy of weekly human serum albumin-paclitaxel nanoparticles in paediatric patients with recurrent or refractory solid tumours included a total of 106 patients aged ≥ 6 months to ≤ 24 years.
The Phase 1 portion of the study included a total of 64 patients aged from 6 months to less than 18 years old and determined the maximum tolerated dose (MTD) to be 240 mg/m², administered as an intravenous infusion over 30 minutes, on Days 1, 8, and 15 of each 28-day cycle.
The Phase 2 portion enrolled a total of 42 patients using a Simon two-stage minimax design, aged from 6 months to 24 years with recurrent or refractory Ewing's sarcoma, neuroblastoma or rhabdomyosarcoma for the evaluation of antitumour activity assessed by the overall response rate (ORR). Of the 42 patients, 1 patient was < 2, 27 were aged ≥ 2 to < 12, 12 were aged ≥12 to < 18 and 2 adult patients were aged ≥ 18 to 24 years old.
Patients were treated for a median of 2 cycles at the MTD. From the 41 patients eligible for efficacy evaluation in stage 1, 1 patient in the rhabdomyosarcoma group (N=14) had a confirmed partial response (PR) resulting in an ORR of 7.1% (95% CI: 0.2, 33.9). No confirmed complete response (CR) or PR was observed in either the Ewing's sarcoma group (N=13) or the neuroblastoma group (N=14). None of the study arms continued into stage 2 because the protocol-defined requirement of ≥ 2 patients to have a confirmed response was not met.
The median overall survival results, including the 1-year follow-up period were 32.1 weeks (95% CI: 21.4, 72.9), 32.0 weeks (95% CI: 12, not established) and 19.6 weeks (95% CI: 4, 25.7) for the Ewing's sarcoma, neuroblastoma and rhabdomyosarcoma groups, respectively.
The overall safety profile of human serum albumin-paclitaxel nanoparticles in paediatric patients was consistent with the known safety profile of human serum albumin-paclitaxel nanoparticles in adults (see section 4.8). Based on these results, it was concluded that human serum albumin-paclitaxel nanoparticles as monotherapy does not have meaningful clinical activity or survival benefit that warrants further development in the paediatric population.
5.2. Pharmacokinetic properties
The pharmacokinetics of total paclitaxel following 30- and 180 minute infusions of human serum albumin-paclitaxel nanoparticles at dose levels of 80 to 375 mg/m² were determined in clinical studies. The paclitaxel exposure (AUC) increased linearly from 2653 to 16736 ng.hr/ml following dosing from 80 to 300 mg/m².
In a study in patients with advanced solid tumours, the pharmacokinetic characteristics of paclitaxel following human serum albumin-paclitaxel nanoparticles administered intravenously at 260 mg/m² over 30 minutes were compared with those following 175 mg/m² of the solvent-based paclitaxel injection administered over 3 hours. Based on non-compartmental PK analysis, the plasma clearance of paclitaxel with human serum albumin-paclitaxel nanoparticles was larger (43%) than that following a solventbased paclitaxel injection and its volume of distribution was also higher (53%). There were no differences in terminal half-lives.
In a repeat dose study with 12 patients receiving human serum albumin-paclitaxel nanoparticles administered intravenously at 260 mg/m², intra-patient variability in AUC was 19% (range = 3.21%- 37.70%). There was no evidence for accumulation of paclitaxel with multiple treatment courses.
Distribution
Following human serum albumin-paclitaxel nanoparticles administration to patients with solid tumours, paclitaxel is evenly distributed into blood cells and plasma and is highly bound to plasma proteins (94%).
The protein binding of paclitaxel following human serum albumin-paclitaxel nanoparticles was evaluated by ultrafiltration in a within-patient comparison study. The fraction of free paclitaxel was significantly higher with human serum albumin-paclitaxel nanoparticles (6.2%) than with solvent-based paclitaxel (2.3%). This resulted in significantly higher exposure to unbound paclitaxel with human serum albumin-paclitaxel nanoparticles compared with solvent-based paclitaxel, even though the total exposure is comparable. This is possibly due to paclitaxel not being trapped in Cremophor EL micelles as with solvent-based paclitaxel. Based on the published literature, in vitro studies of binding to human serum proteins, (using paclitaxel at concentrations ranging from 0.1 to 50 µg/ml), indicate that the presence of cimetidine, ranitidine, dexamethasone, or diphenhydramine did not affect protein binding of paclitaxel.
Based on population pharmacokinetic analysis, the total volume of distribution is approximately 1741 L; the large volume of distribution indicates extensive extravascular distribution and/or tissue binding of paclitaxel.
Biotransformation and elimination
Based on the published literature, in vitro studies with human liver microsomes and tissue slices show that paclitaxel is metabolised primarily to 6α-hydroxypaclitaxel; and to two minor metabolites, 3'-phydroxypaclitaxel and 6α-3'-p-dihydroxypaclitaxel. The formation of these hydroxylated metabolites is catalysed by CYP2C8, CYP3A4, and both CYP2C8 and CYP3A4 isoenzymes, respectively.
In patients with metastatic breast cancer, after a 30-minute infusion of human serum albumin-paclitaxel nanoparticles at 260 mg/m² , the mean value for cumulative urinary excretion of unchanged active substance accounted for 4% of the total administered dose with less than 1% as the metabolites 6αhydroxypaclitaxel and 3'-p-hydroxypaclitaxel, indicating extensive non-renal clearance. Paclitaxel is principally eliminated by hepatic metabolism and biliary excretion.
At the clinical dose range of 80 to 300 mg/m², the mean plasma clearance of paclitaxel ranges from 13 to 30 L/h/m², and the mean terminal half-life ranges from 13 to 27 hours.
Hepatic impairment
The effect of hepatic impairment on population pharmacokinetics of human serum albumin-paclitaxel nanoparticles was studied in patients with advanced solid tumours. This analysis included patients with normal hepatic function (n=130), and pre-existing mild (n=8), moderate (n=7), or severe (n=5) hepatic impairment (according to NCI Organ Dysfunction Working Group criteria). The results show that mild hepatic impairment (total bilirubin >1 to ≤1.5 x ULN) has no clinically important effect on pharmacokinetics of paclitaxel. Patients with moderate (total bilirubin >1.5 to ≤3 x ULN) or severe (total bilirubin >3 to ≤5 x ULN) hepatic impairment have a 22% to 26% decrease in the maximum elimination rate of paclitaxel and approximately 20% increase in mean paclitaxel AUC compared with patients with normal hepatic function. Hepatic impairment has no effect on mean paclitaxel Cmax. In addition, elimination of paclitaxel shows an inverse correlation with total bilirubin and a positive correlation with serum albumin.
Pharmacokinetic/pharmacodynamic modeling indicates that there is no correlation between hepatic function (as indicated by the baseline albumin or total bilirubin level) and neutropenia after adjusting for human serum albumin-paclitaxel nanoparticles exposure.
Pharmacokinetic data are not available for patients with total bilirubin >5 x ULN or for patients with metastatic adenocarcinoma of the pancreas (see section 4.2).
Renal impairment
Population pharmacokinetic analysis included patients with normal renal function (n=65), and preexisting mild (n=61), moderate (n=23), or severe (n=l) renal impairment (according to draft FDA guidance criteria 2010). Mild to moderate renal impairment (creatinine clearance ≥30 to <90 ml/min) has no clinically important effect on the maximum elimination rate and systemic exposure (AUC and Cmax) of paclitaxel. Pharmacokinetic data are insufficient for patients with severe renal impairment and not available for patients with end stage kidney disease.
Elderly
Population pharmacokinetic analysis for human serum albumin-paclitaxel nanoparticles included patients with ages ranging from 24 to 85 years old and shows that age does not significantly influence the maximum elimination rate and systemic exposure (AUC and Cmax) of paclitaxel.
Pharmacokinetic/pharmacodynamic modelling using data from 125 patients with advanced solid tumours indicates that patients ≥ 65 years of age may be more susceptible to development of neutropenia within the first treatment cycle, although the plasma paclitaxel exposure is not affected by age.
Paediatric population
The pharmacokinetics of paclitaxel following 30 minutes of intravenous administration at dose levels of 120 mg/m² to 270 mg/m² were determined in 64 patients (2 to ≤ 18 years) in Phase 1 of a Phase ½ study in recurrent or refractory paediatric solid tumours. Following dosing increase from 120 to 270 mg/m², the paclitaxel mean AUC(0-inf) and Cmax ranged from 8867 to 14361 ng*hr/ml and from 3488 to 8078 ng/ml, respectively.
Dose normalized peak drug exposure values were comparable across the dose range studied; however, dose-normalized total drug exposure values were only comparable across 120 mg/m² to 240 mg/m²; with lower dose-normalized AUC∞ at the 270 mg/m² dose level. At the MTD of 240 mg/m², the mean CL was 19.1 L/h and the mean terminal half-life was 13.5 hours.
In children and adolescent patients, exposure to paclitaxel increased with higher dosing and weekly drug exposures were higher than in adult patients.
Other intrinsic factors
Population pharmacokinetic analyses for human serum albumin-paclitaxel nanoparticles indicate that gender, race (Asian vs. White), and type of solid tumours do not have a clinically important effect on systemic exposure (AUC and Cmax) of paclitaxel. Patients weighing 50 kg had paclitaxel AUC approximately 25% lower than those weighing 75 kg. The clinical relevance of this finding is uncertain.
5.3. Preclinical safety data
The carcinogenic potential of paclitaxel has not been studied. However, based on the published literature, paclitaxel is a potentially carcinogenic and genotoxic agent at clinical doses, based upon its pharmacodynamic mechanism of action. Paclitaxel has been shown to be clastogenic in vitro (chromosome aberrations in human lymphocytes) and in vivo (micronucleus test in mice). Paclitaxel has been shown to be genotoxic in vivo (micronucleus test in mice), but it did not induce mutagenicity in the Ames test or the Chinese hamster ovary/hypoxanthine-guanine phosphoribosyl transferase (CHO/HGPRT) gene mutation assay.
Paclitaxel at doses below the human therapeutic dose was associated with low fertility when administered prior and during mating in male and female rats and foetal toxicity in rats. Animal studies with human serum albumin-paclitaxel nanoparticles showed non-reversible, toxic effects on the male reproductive organs at clinically relevant exposure levels.
Paclitaxel and/or its metabolites were excreted into the milk of lactating rats. Following intravenous administration of radiolabelled paclitaxel to rats on days 9 to 10 postpartum, concentrations of radioactivity in milk were higher than in plasma and declined in parallel with the plasma concentrations.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.