ODEFSEY Film-coated tablet Ref.[28335] Active ingredients: Emtricitabine Rilpivirine Tenofovir alafenamide
Source: European Medicines Agency (EU) Revision Year: 2021 Publisher: Gilead Sciences Ireland UC, Carrigtohill, County Cork, T45 DP77, Ireland
4.3. Contraindications
Hypersensitivity to the active substances or to any of the excipients listed in section 6.1.
Odefsey should not be co-administered with medicinal products that can result in significant decreases in rilpivirine plasma concentrations (due to cytochrome P450 [CYP]3A enzyme induction or gastric pH increase), which may result in loss of therapeutic effect of Odefsey (see section 4.5), including:
- carbamazepine, oxcarbazepine, phenobarbital, phenytoin
- rifabutin, rifampicin, rifapentine
- omeprazole, esomeprazole, dexlansoprazole, lansoprazole, pantoprazole, rabeprazole
- dexamethasone (oral and parenteral doses), except as a single dose treatment
- St. John’s wort (Hypericum perforatum)
4.4. Special warnings and precautions for use
While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of sexual transmission, a residual risk cannot be excluded. Precautions to prevent transmission should be taken in accordance with national guidelines.
Virologic failure and development of resistance
There are insufficient data to justify the use in patients with prior NNRTI failure. Resistance testing and/or historical resistance data should guide the use of Odefsey (see section 5.1).
In the pooled efficacy analysis from the two Phase 3 clinical studies in adults (C209 [ECHO] and C215 [THRIVE]) through 96 weeks, patients treated with emtricitabine/tenofovir disoproxil fumarate + rilpivirine with a baseline viral load >100,000 HIV-1 RNA copies/mL had a greater risk of virologic failure (17.6% with rilpivirine versus 7.6% with efavirenz) compared to patients with a baseline viral load ≤100,000 HIV-1 RNA copies/mL (5.9% with rilpivirine versus 2.4% with efavirenz). The virologic failure rate in patients treated with emtricitabine/tenofovir disoproxil fumarate + rilpivirine at Week 48 and Week 96 was 9.5% and 11.5% respectively, and 4.2% and 5.1% in the emtricitabine/tenofovir disoproxil fumarate + efavirenz arm. The difference in the rate of new virologic failures from the Week 48 to Week 96 analysis between rilpivirine and efavirenz arms was not statistically significant. Patients with a baseline viral load >100,000 HIV-1 RNA copies/mL who experienced virologic failure exhibited a higher rate of treatment-emergent resistance to the NNRTI class. More patients who failed virologically on rilpivirine than who failed virologically on efavirenz developed lamivudine/emtricitabine associated resistance (see section 5.1).
Findings in adolescents (12 to less than 18 years of age) in Study C213 were generally in line with these data (for details see section 5.1).
Only adolescents deemed likely to have good adherence to antiretroviral therapy should be treated with rilpivirine, as suboptimal adherence can lead to development of resistance and the loss of future treatment options.
Cardiovascular
At supratherapeutic doses (75 mg once daily and 300 mg once daily), rilpivirine has been associated with prolongation of the QTc interval of the electrocardiogram (ECG) (see sections 4.5 and 4.9). Rilpivirine at the recommended dose of 25 mg once daily is not associated with a clinically relevant effect on QTc. Odefsey should be used with caution when co-administered with medicinal products with a known risk of Torsade de Pointes.
Patients co-infected with HIV and hepatitis B or C virus
Patients with chronic hepatitis B or C treated with antiretroviral therapy are at an increased risk for severe and potentially fatal hepatic adverse reactions.
The safety and efficacy of Odefsey in patients co-infected with HIV-1 and hepatitis C virus (HCV) have not been established.
Tenofovir alafenamide is active against hepatitis B virus (HBV). Discontinuation of Odefsey therapy in patients co-infected with HIV and HBV may be associated with severe acute exacerbations of hepatitis. Patients co-infected with HIV and HBV who discontinue Odefsey should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment.
Liver disease
The safety and efficacy of Odefsey in patients with significant underlying liver disorders have not been established.
Patients with pre-existing liver dysfunction, including chronic active hepatitis, have an increased frequency of liver function abnormalities during combination antiretroviral therapy (CART) and should be monitored according to standard practice. If there is evidence of worsening liver disease in such patients, interruption or discontinuation of treatment must be considered.
Weight and metabolic parameters
An increase in weight and in levels of blood lipids and glucose may occur during antiretroviral therapy. Such changes may in part be linked to disease control and lifestyle. For lipids, there is in some cases evidence for a treatment effect, while for weight gain there is no strong evidence relating this to any particular treatment. For monitoring of blood lipids and glucose reference is made to established HIV treatment guidelines. Lipid disorders should be managed as clinically appropriate.
Mitochondrial dysfunction following exposure in utero
Nucleos(t)ide analogues may impact mitochondrial function to a variable degree, which is most pronounced with stavudine, didanosine and zidovudine. There have been reports of mitochondrial dysfunction in HIV negative infants exposed in utero and/or postnatally to nucleoside analogues; these have predominantly concerned treatment with regimens containing zidovudine. The main adverse reactions reported are haematological disorders (anaemia, neutropenia) and metabolic disorders (hyperlactatemia, hyperlipasemia). These events have often been transitory. Late onset neurological disorders have been reported rarely (hypertonia, convulsion, abnormal behaviour). Whether such neurological disorders are transient or permanent is currently unknown. These findings should be considered for any child exposed in utero to nucleos(t)ide analogues, who present with severe clinical findings of unknown aetiology, particularly neurologic findings. These findings do not affect current national recommendations to use antiretroviral therapy in pregnant women to prevent vertical transmission of HIV.
Immune Reactivation Syndrome
In HIV infected patients with severe immune deficiency at the time of institution of CART, an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of CART. Relevant examples include cytomegalovirus retinitis, generalised and/or focal mycobacterial infections, and Pneumocystis jirovecii pneumonia. Any inflammatory symptoms should be evaluated and treatment instituted when necessary.
Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment.
Opportunistic infections
Patients receiving Odefsey may continue to develop opportunistic infections and other complications of HIV infection, and therefore should remain under close clinical observation by physicians experienced in the treatment of patients with HIV associated diseases.
Osteonecrosis
Although the aetiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV disease and/or long-term exposure to CART. Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
Nephrotoxicity
A potential risk of nephrotoxicity resulting from chronic exposure to low levels of tenofovir due to dosing with tenofovir alafenamide cannot be excluded (see section 5.3).
It is recommended that renal function is assessed in all patients prior to, or when initiating, therapy with Odefsey and that it is also monitored during therapy in all patients as clinically appropriate. In patients who develop clinically significant decreases in renal function, or evidence of proximal renal tubulopathy, discontinuation of Odefsey should be considered.
Patients with end stage renal disease on chronic haemodialysis
Odefsey should generally be avoided but may be used with caution in adults with end stage renal disease (estimated CrCl <15 mL/min) on chronic haemodialysis if the potential benefits outweigh the potential risks (see section 4.2). In a study of emtricitabine + tenofovir alafenamide in combination with elvitegravir + cobicistat as a fixed-dose combination tablet (E/C/F/TAF) in HIV-1 infected adults with end stage renal disease (estimated CrCl <15 mL/min) on chronic haemodialysis, efficacy was maintained through 48 weeks but emtricitabine exposure was significantly higher than in patients with normal renal function. Although there were no new safety issues identified, the implications of increased emtricitabine exposure remain uncertain (see sections 4.8 and 5.2).
Pregnancy
Lower exposures of rilpivirine were observed when rilpivirine 25 mg once daily was taken during pregnancy. In the Phase 3 studies (C209 and C215), lower rilpivirine exposure, similar to that seen during pregnancy, has been associated with an increased risk of virological failure, therefore viral load should be monitored closely (see sections 4.6, 5.1 and 5.2). Alternatively, switching to another antiretroviral regimen could be considered.
Co-administration of other medicinal products
Some medicinal products should not be co-administered with Odefsey (see sections 4.3 and 4.5).
Odefsey should not be co-administered with other antiretroviral medicinal products (see section 4.5).
Odefsey should not be co-administered with other medicinal products containing tenofovir alafenamide, lamivudine, tenofovir disoproxil or adefovir dipivoxil (see section 4.5).
Excipients
Odefsey contains lactose monohydrate. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take this medicinal product.
This medicine contains less than 1 mmol sodium (23 mg) per tablet, that is to say essentially ‘sodiumfree’.
4.5. Interaction with other medicinal products and other forms of interaction
Odefsey is indicated for use as a complete regimen for the treatment of HIV-1 infection and should not be co-administered with other antiretroviral medicinal products. Therefore, information regarding drug-drug interactions with other antiretroviral medicinal products is not provided. Interaction studies have only been performed in adults.
Emtricitabine
In vitro and clinical pharmacokinetic drug-drug interaction studies have shown that the potential for CYP-mediated interactions involving emtricitabine with other medicinal products is low. Co-administration of emtricitabine with medicinal products that are eliminated by active tubular secretion may increase concentrations of emtricitabine, and/or the co-administered medicinal product. Medicinal products that decrease renal function may increase concentrations of emtricitabine.
Rilpivirine
Rilpivirine is primarily metabolised by CYP3A. Medicinal products that induce or inhibit CYP3A may thus affect the clearance of rilpivirine (see section 5.2). Rilpivirine inhibits P-glycoprotein (P-gp) in vitro (50% inhibitory concentration [IC50] is 9.2 µM). In a clinical study, rilpivirine did not significantly affect the pharmacokinetics of digoxin. Additionally, in a clinical drug-drug interaction study with tenofovir alafenamide, which is more sensitive to intestinal P-gp inhibition, rilpivirine did not affect tenofovir alafenamide exposures when administered concurrently, indicating that rilpivirine is not a P-gp inhibitor in vivo.
Rilpivirine is an in vitro inhibitor of the transporter MATE-2K with an IC50 of < 2.7 nM. The clinical implications of this finding are currently unknown.
Tenofovir alafenamide
Tenofovir alafenamide is transported by P-gp and breast cancer resistance protein (BCRP). Medicinal products that affect P-gp and BCRP activity may lead to changes in tenofovir alafenamide absorption (see Table 1). Medicinal products that induce P-gp activity (e.g., rifampicin, rifabutin, carbamazepine, phenobarbital) are expected to decrease the absorption of tenofovir alafenamide, resulting in decreased plasma concentration of tenofovir alafenamide, which may lead to loss of therapeutic effect of Odefsey and development of resistance. Co-administration of Odefsey with other medicinal products that inhibit P-gp and BCRP activity (e.g., ketoconazole, fluconazole, itraconazole, posaconazole, voriconazole, ciclosporin) is expected to increase the absorption and plasma concentration of tenofovir alafenamide. Based on data from an in vitro study, co-administration of tenofovir alafenamide and xanthine oxidase inhibitors (e.g., febuxostat) is not expected to increase systemic exposure to tenofovir in vivo.
Tenofovir alafenamide is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 or CYP2D6 in vitro. Tenofovir alafenamide is not an inhibitor or inducer of CYP3A in vivo. Tenofovir alafenamide is a substrate of organic anion transporting polypeptide (OATP) 1B1 and OATP1B3 in vitro. The distribution of tenofovir alafenamide in the body may be affected by the activity of OATP1B1 and OATP1B3.
Concomitant use contraindicated
Co-administration of Odefsey and medicinal products that induce CYP3A has been observed to decrease the plasma concentrations of rilpivirine which could potentially lead to loss of virologic response to Odefsey (see section 4.3) and possible resistance to rilpivirine and to the NNRTI class.
Co-administration of Odefsey with proton pump inhibitors has been observed to decrease the plasma concentrations of rilpivirine (due to an increase in gastric pH) which could potentially lead to loss of virologic response to Odefsey (see section 4.3) and possible resistance to rilpivirine and to the NNRTI class.
Concomitant use where caution is recommended
CYP enzyme inhibitors
Co-administration of Odefsey with medicinal products that inhibit CYP3A enzyme activity has been observed to increase rilpivirine plasma concentrations.
QT prolonging medicinal products
Odefsey should be used with caution when co-administered with a medicinal product with a known risk of Torsade de Pointes (see section 4.4).
Other interactions
Tenofovir alafenamide is not an inhibitor of human uridine diphosphate glucuronosyltransferase (UGT) 1A1 in vitro. It is not known whether emtricitabine, or tenofovir alafenamide are inhibitors of other UGT enzymes. Emtricitabine did not inhibit the glucuronidation reaction of a non-specific UGT substrate in vitro.
Interactions between Odefsey or its individual component(s) and co-administered medicinal products are listed in Table 1 below (increase is indicated as “↑”, decrease as “↓” and no change as “↔”).
Table 1. Interactions between Odefsey or its individual component(s) and other medicinal products:
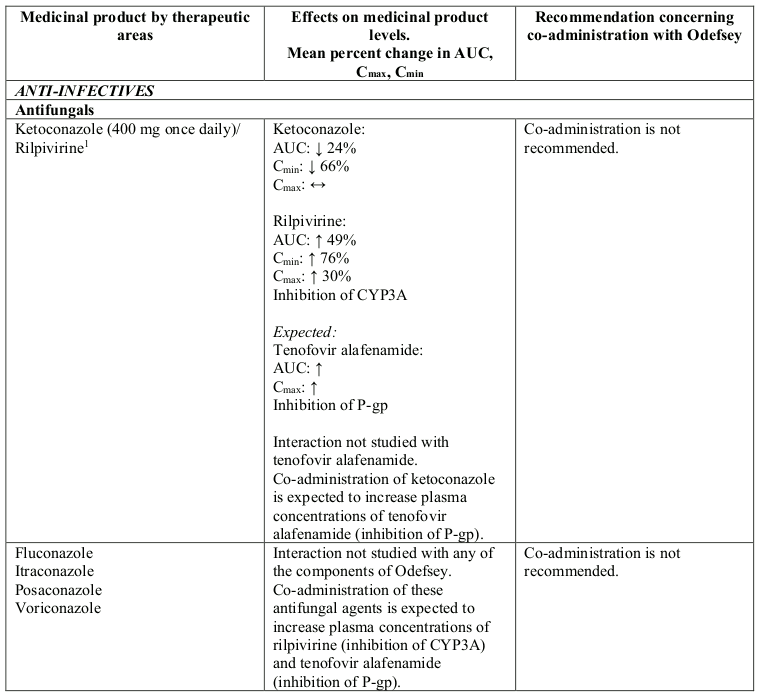



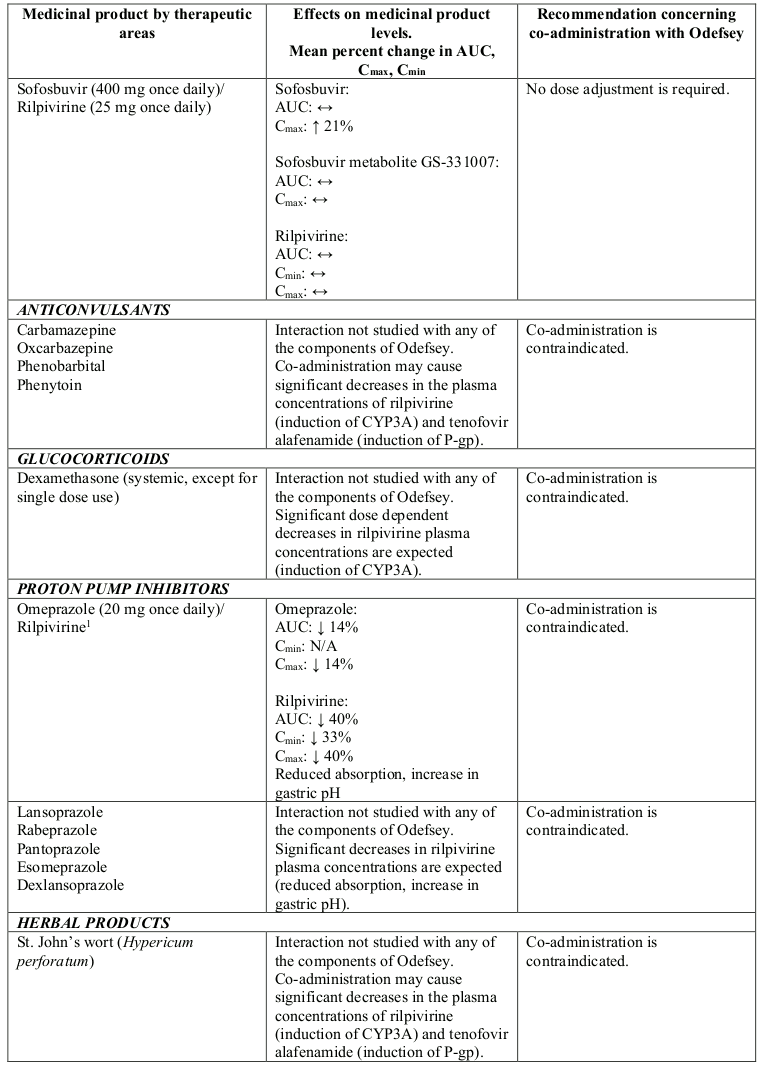
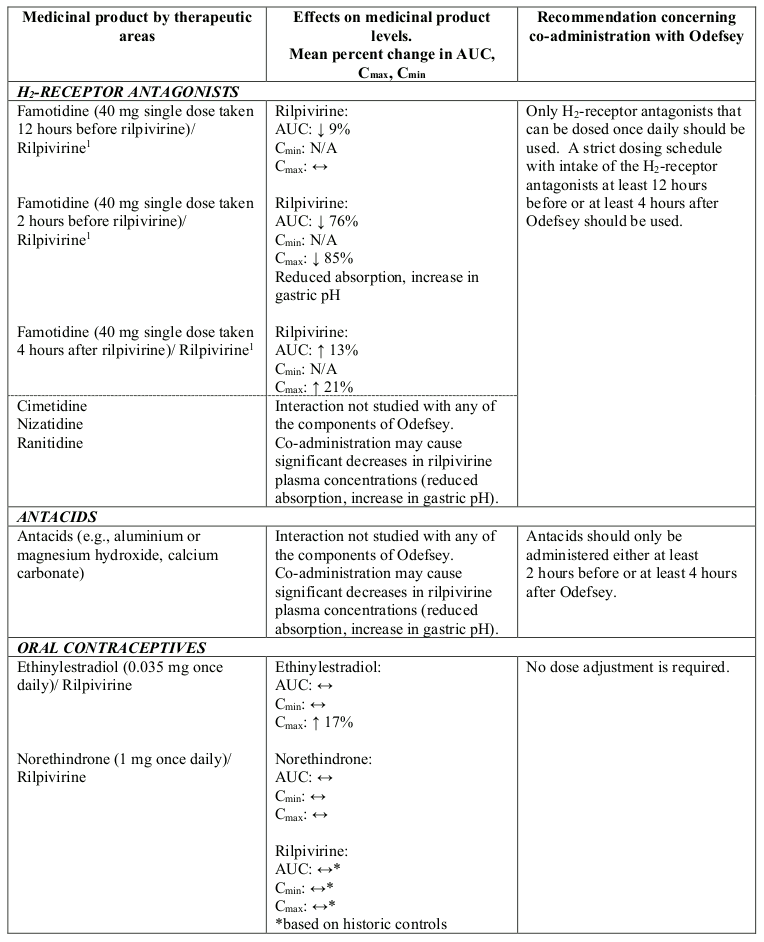
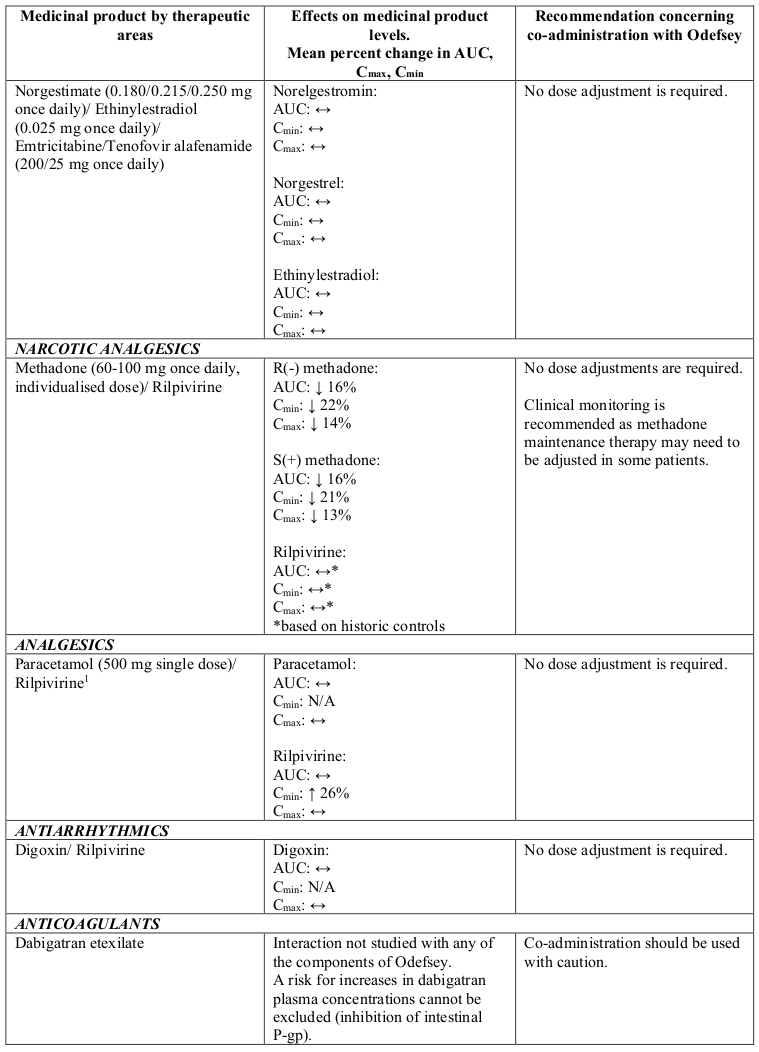
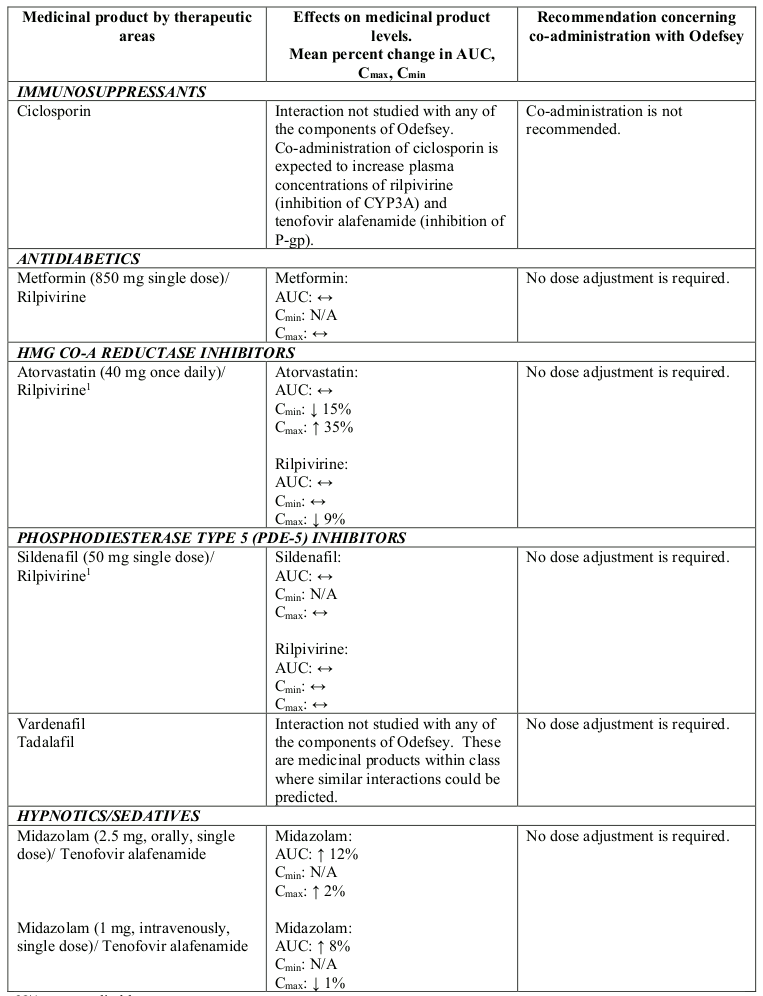
N/A = not applicable
1 This interaction study has been performed with a dose higher than the recommended dose for rilpivirine hydrochloride assessing the maximal effect on the co-administered medicinal product. The dosing recommendation is applicable to the recommended dose of rilpivirine of 25 mg once daily.
2 Study conducted with emtricitabine/rilpivirine/tenofovir disoproxil fumarate fixed-dose combination tablet.
3 Study conducted with additional voxilaprevir 100 mg to achieve voxilaprevir exposures expected in HCV infected patients.
Studies conducted with other medicinal products
Based on drug-drug interaction studies conducted with the components of Odefsey, no clinically significant interactions are expected when Odefsey is combined with the following medicinal products: buprenorphine, naloxone and norbuprenorphine.
4.6. Fertility, pregnancy and lactation
Women of childbearing potential/contraception in males and females
The use of Odefsey should be accompanied by the use of effective contraception.
Pregnancy
There are no adequate and well-controlled studies of Odefsey or its components in pregnant women.
There is a limited amount of data (less than 300 pregnancy outcomes) from the use of tenofovir alafenamide in pregnant women. A moderate amount of data on pregnant women (between 300-1,000 pregnancy outcomes) indicate no malformative or foetal/neonatal toxicity of rilpivirine (see sections 4.4, 5.1 and 5.2). Lower exposures of rilpivirine were observed during pregnancy; therefore viral load should be monitored closely. A large amount of data on pregnant women (more than 1,000 exposed outcomes) indicate no malformative nor foetal/neonatal toxicity associated with emtricitabine.
Animal studies do not indicate direct or indirect harmful effects with respect to reproductive toxicity (see section 5.3) with the components of Odefsey.
Odefsey should be used during pregnancy only if the potential benefit justifies the potential risk to the foetus.
Breast-feeding
Emtricitabine is excreted in human milk. It is not known whether rilpivirine or tenofovir alafenamide are excreted in human milk. In animal studies it has been shown that tenofovir is excreted in milk. Rilpivirine is excreted in the milk of rats.
There is insufficient information on the effects of all the components of Odefsey in newborns/infants.
Because of both the potential for HIV transmission and the potential for adverse reactions in breastfed infants, women should be instructed not to breast-feed if they are receiving Odefsey.
Fertility
No human data on the effect of Odefsey on fertility are available. Animal studies do not indicate harmful effects of emtricitabine, rilpivirine hydrochloride or tenofovir alafenamide on fertility (see section 5.3).
4.7. Effects on ability to drive and use machines
Odefsey may have minor influence on the ability to drive and use machines. Patients should be informed that fatigue, dizziness and somnolence have been reported during treatment with the components of Odefsey (see section 4.8). This should be considered when assessing a patient’s ability to drive or operate machinery.
4.8. Undesirable effects
Summary of the safety profile
The most frequently reported adverse reactions in clinical studies of treatment-naïve patients taking emtricitabine + tenofovir alafenamide in combination with elvitegravir + cobicistat were nausea (11%), diarrhoea (7%), and headache (6%). The most frequently reported adverse reactions in clinical studies of treatment-naïve patients taking rilpivirine hydrochloride in combination with emtricitabine + tenofovir disoproxil fumarate were nausea (9%), dizziness (8%), abnormal dreams (8%), headache (6%), diarrhoea (5%) and insomnia (5%).
Tabulated summary of adverse reactions
Assessment of adverse reactions is based on safety data from across all Phase 2 and 3 studies in which patients received emtricitabine + tenofovir alafenamide given with elvitegravir + cobicistat as a fixed-dose combination tablet, pooled data from patients who received rilpivirine 25 mg once daily in combination with other antiretroviral medicinal products in the controlled studies TMC278-C209 and TMC278-C215, patients who received Odefsey in Studies GS-US-366-1216 and GS-US-366-1160, and post-marketing experience.
The adverse reactions in Table 2 are listed by system organ class and highest frequency observed. Frequencies are defined as follows: very common (≥1/10), common (≥1/100 to <1/10) or uncommon (≥1/1,000 to <1/100).
Table 2. Tabulated list of adverse reactions:
| Frequency | Adverse reaction |
|---|---|
| Blood and lymphatic system disorders | |
| Common: | decreased white blood cell count1, decreased haemoglobin1, decreased platelet count1Uncommon: anaemia2 |
| Immune system disorders | |
| Uncommon: | immune reactivation syndrome1 |
| Metabolism and nutrition disorders | |
| Very common: | increased total cholesterol (fasted)1, increased LDL-cholesterol (fasted)1 |
| Common: | decreased appetite1, increased triglycerides (fasted)1 |
| Psychiatric disorders | |
| Very common: | insomnia1 |
| Common: | depression1, abnormal dreams1,3, sleep disorders1, depressed mood1 |
| Nervous system disorders | |
| Very common: | headache1,3, dizziness1,3 |
| Common: somnolence1 | |
| Gastrointestinal disorders | |
| Very common: | nausea1,3, increased pancreatic amylase1 |
| Common: | abdominal pain1,3, vomiting1,3, increased lipase1, abdominal discomfort1, dry mouth1, flatulence3, diarrhoea3 |
| Uncommon: | dyspepsia3 |
| Hepatobiliary disorders | |
| Very common: | increased transaminases (AST and/or ALT)1 |
| Common: | increased bilirubin1 |
| Skin and subcutaneous tissue disorders | |
| Common: | rash1,3 |
| Uncommon: | severe skin reactions with systemic symptoms4, angioedema5,6, pruritus3, urticaria6 |
| Musculoskeletal and connective tissue disorders | |
| Uncommon: | arthralgia3 |
| General disorders and administration site conditions | |
| Common: | fatigue1,3 |
1 Adverse reactions identified from rilpivirine clinical studies.
2 This adverse reaction was not observed in the Phase 3 studies of emtricitabine + tenofovir alafenamide in combination with elvitegravir + cobicistat or in the Phase 3 studies with Odefsey but identified from clinical studies or post-marketing experience of emtricitabine when used with other antiretrovirals.
3 Adverse reactions identified from clinical studies of emtricitabine + tenofovir alafenamide-containing products.
4 Adverse reaction identified through post-marketing surveillance of emtricitabine/rilpivirine/tenofovir disoproxil fumarate.
5 Adverse reaction identified through post-marketing surveillance for emtricitabine-containing products.
6 Adverse reaction identified through post-marketing surveillance for tenofovir alafenamide-containing products.
Laboratory abnormalities
Changes in serum creatinine for rilpivirine-containing regimens
The pooled data from the Phase 3 TMC278-C209 and TMC278-C215 studies of treatment-naïve patients also demonstrate that serum creatinine increased and estimated glomerular filtration rate (eGFR) decreased over 96 weeks of treatment with rilpivirine. Most of this increase in creatinine and decrease in eGFR occurred within the first four weeks of treatment. Over 96 weeks of treatment with rilpivirine mean changes of 0.1 mg/dL (range: -0.3 mg/dL to 0.6 mg/dL) for creatinine and -13.3 mL/min/1.73 m² (range: -63.7 mL/min/1.73 m² to 40.1 mL/min/1.73 m²) for eGFR were observed. In patients who entered the studies with mild or moderate renal impairment, the serum creatinine increase observed was similar to that seen in patients with normal renal function. These increases do not reflect a change in actual glomerular filtration rate (GFR).
Changes in lipid laboratory tests
In studies in treatment-naïve patients receiving emtricitabine + tenofovir alafenamide (FTC + TAF) or emtricitabine + tenofovir disoproxil fumarate (FTC + TDF), both given with elvitegravir + cobicistat as a fixed-dose combination tablet, increases from baseline were observed in both treatment groups for the fasting lipid parameters total cholesterol, direct low-density lipoprotein (LDL)- and high-density lipoprotein (HDL)cholesterol, and triglycerides at Week 144. The median increase from baseline for these parameters was greater in patients receiving FTC + TAF compared with patients receiving FTC + TDF (p<0.001 for the difference between treatment groups for fasting total cholesterol, direct LDL and HDL-cholesterol, and triglycerides). Median (Q1, Q3) change from baseline at Week 144 in total cholesterol to HDL-cholesterol ratio was 0.2 (-0.3, 0.7) in patients receiving FTC + TAF and 0.1 (-0.4, 0.6) in patients receiving FTC + TDF (p=0.006 for the difference between treatment groups).
Switching from a TDF-based regimen to Odefsey may lead to slight increases in lipid parameters. In a study of virologically suppressed patients switching from FTC/RPV/TDF to Odefsey (Study GS-US-366-1216), increases from baseline were observed in fasting values of total cholesterol, direct LDL-cholesterol, HDL-cholesterol, and triglycerides in the Odefsey arm; and no clinically relevant changes from baseline in median fasting values for total cholesterol to HDL-cholesterol ratio were observed in either treatment arm at Week 96. In a study of virologically suppressed patients switching from EFV/FTC/TDF to Odefsey (Study GS-US-366-1160), decreases from baseline were observed in the fasting values of total cholesterol and HDL-cholesterol in the Odefsey arm; no clinically relevant changes from baseline in median fasting values for total cholesterol to HDL-cholesterol ratio, direct LDL-cholesterol or triglycerides were observed in either treatment arm at Week 96.
Cortisol
In the pooled Phase 3 TMC278-C209 and TMC278-C215 studies of treatment-naïve patients, at Week 96, there was an overall mean change from baseline in basal cortisol of -19.1 (-30.85; -7.37) nmol/L in the rilpivirine arm and of -0.6 (-13.29; 12.17) nmol/L in the efavirenz arm. At Week 96, the mean change from baseline in ACTH-stimulated cortisol levels was lower in the rilpivirine arm (+18.4 ± 8.36 nmol/L) than in the efavirenz arm (+54.1 ± 7.24 nmol/L). Mean values for the rilpivirine arm for both basal and ACTH-stimulated cortisol at Week 96 were within the normal range. These changes in adrenal safety parameters were not clinically relevant. There were no clinical signs or symptoms suggestive of adrenal or gonadal dysfunction in adults.
Description of selected adverse reactions
Metabolic parameters
Weight and levels of blood lipids and glucose may increase during antiretroviral therapy (see section 4.4).
Immune Reactivation Syndrome
In HIV infected patients with severe immune deficiency at the time of initiation of CART, an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
Osteonecrosis
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to CART. The frequency of this is unknown (see section 4.4).
Severe skin reactions
Severe skin reactions with systemic symptoms have been reported during post-marketing experience of emtricitabine/rilpivirine/tenofovir disoproxil fumarate including rashes accompanied by fever, blisters, conjunctivitis, angioedema, elevated liver function tests, and/or eosinophilia.
Paediatric population
The safety of emtricitabine + tenofovir alafenamide was evaluated through 48 weeks in an open-label clinical study (GS-US-292-0106) in which 50 HIV-1 infected, treatment-naïve paediatric patients aged 12 to <18 years received emtricitabine + tenofovir alafenamide in combination with elvitegravir + cobicistat as a fixed-dose combination tablet. In this study, the safety profile in adolescent patients was similar to that in adults (see section 5.1).
The safety assessment of rilpivirine is based on Week 48 data from one single-arm open-label study (TMC278-C213) in 36 paediatric patients 12 to <18 years and weighing at least 32 kg. No patients discontinued rilpivirine due to adverse reactions. No new adverse reactions were identified compared to those seen in adults. Most adverse reactions were Grade 1 or 2. Adverse reactions (all grades) of very common frequency were headache, depression, somnolence and nausea. No Grade 3-4 laboratory abnormalities for AST/ALT or Grade 3-4 adverse reactions of transaminase increased were reported (see section 5.1).
Other special populations
Patients with renal impairment
The safety of emtricitabine + tenofovir alafenamide was evaluated through 144 weeks in an open-label clinical study (GS-US-292-0112), in which 248 HIV-1 infected patients who were either treatment-naïve (n=6) or virologically suppressed (n=242) with mild to moderate renal impairment (estimated glomerular filtration rate by Cockcroft-Gault method [eGFRCG]: 30-69 mL/min) received emtricitabine + tenofovir alafenamide in combination with elvitegravir + cobicistat as a fixed-dose combination tablet. The safety profile in patients with mild to moderate renal impairment was similar to that in patients with normal renal function (see section 5.1).
The safety of emtricitabine + tenofovir alafenamide was evaluated through 48 weeks in a single arm, open-label clinical study (GS-US-292-1825) in which 55 virologically suppressed HIV-1 infected patients with end stage renal disease (eGFRCG <15 mL/min) on chronic haemodialysis received emtricitabine + tenofovir alafenamide in combination with elvitegravir + cobicistat as a fixed-dose combination tablet. There were no new safety issues identified in patients with end stage renal disease on chronic haemodialysis receiving emtricitabine + tenofovir alafenamide, given with elvitegravir + cobicistat as a fixed-dose combination tablet (see section 5.2).
Patients co-infected with HIV and HBV
The safety of emtricitabine + tenofovir alafenamide in combination with elvitegravir and cobicistat as a fixed-dose combination tablet (elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide [E/C/F/TAF]) was evaluated in 72 HIV/HBV co-infected patients receiving treatment for HIV in an open-label clinical study (GS-US-292-1249), through Week 48, in which patients were switched from another antiretroviral regimen (which included TDF in 69 of 72 patients) to E/C/F/TAF. Based on these limited data, the safety profile of emtricitabine + tenofovir alafenamide in combination with elvitegravir and cobicistat as a fixed-dose combination tablet, in patients with HIV/HBV co-infection, was similar to that in patients with HIV-1 monoinfection.
In patients co-infected with hepatitis B or C virus receiving rilpivirine, the incidence of hepatic enzyme elevation was higher than in patients receiving rilpivirine who were not co-infected. The pharmacokinetic exposure of rilpivirine in co-infected patients was comparable to that in patients without co-infection.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
6.2. Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.