QALSODY Solution for injection Ref.[107302] Active ingredients: Tofersen
Source: FDA, National Drug Code (US) Revision Year: 2023
12.1. Mechanism of Action
Tofersen is an antisense oligonucleotide that causes degradation of SOD1 mRNA through binding to SOD1 mRNA, which results in a reduction of SOD1 protein synthesis.
12.2. Pharmacodynamics
Effect of Tofersen on Total CSF SOD1 Protein
Total CSF SOD1, an indirect measure of target engagement, was evaluated in Study 1 Part C in SOD1-ALS patients [see Clinical Studies (14)].
At Week 28 in Study 1 Part C, a reduction in total CSF SOD1 protein of 35% (geometric mean ratio to baseline) in the tofersen-treated group versus a 2% decrease from baseline in the corresponding placebo subjects in the ITT population was observed (difference in geometric mean ratios for tofersen to placebo: 34%; nominal p<0.0001).
Effect of Tofersen on Neurofilament Proteins
Plasma NfL, a blood-based biomarker of axonal injury and neurodegeneration, was evaluated in Study 1 Part C in SOD1-ALS patients [see Clinical Studies (14)].
At Week 28 in Study 1 Part C, mean plasma NfL was reduced 55% (geometric mean ratio to baseline) in the QALSODY-treated subjects, compared to a 12% increase with placebo in ITT population (difference in geometric mean ratios for QALSODY to placebo: 60%; nominal p<0.0001). Plasma NfL declined until approximately Day 113, after which the reductions were sustained. The reductions in phosphorylated neurofilament heavy chain (pNfH) were similar compared to reductions in NfL, as were reductions in CSF compared to plasma.
Cardiac Electrophysiology
At the maximum approved recommended dosing regimen, QALSODY does not prolong the QTc interval to any clinically relevant extent.
12.3. Pharmacokinetics
Absorption
Intrathecal administration of QALSODY into the CSF allows tofersen to be distributed from the CSF to central nervous system tissues. The maximum CSF trough concentration occurred at the third dose, which was the last dose of the loading period. There was little to no accumulation for CSF tofersen with monthly dosing after the loading phase. Tofersen is transferred from CSF into the systemic circulation, with median time to maximum concentration (Tmax) plasma values ranging from 2 to 6 hours. There was no accumulation in plasma tofersen exposure following monthly maintenance dosing.
Distribution
Autopsy tissue from patients treated with tofersen (n=3) showed that tofersen administered intrathecally was distributed within the central nervous system tissues.
Elimination
Metabolism
Tofersen is expected to be metabolized through exonuclease (3'- and 5')-mediated hydrolysis and is not a substrate for, or inhibitor or inducer of CYP450 enzymes.
Excretion
The primary route of elimination has not been characterized. The effective half-life in CSF is estimated to be 4 weeks.
Drug Interaction Studies
No clinical drug interaction studies have been performed. In vitro, QALSODY is not a substrate or inhibitor/inducer of major CYP enzymes or a substrate or inhibitor of major transporters.
Specific Populations
Effect of sex, race, age, and body weight on tofersen exposure in plasma was not clinically significant. The effect of these factors on tofersen exposure in CSF is unknown.
Patients with Renal or Hepatic Impairment
No clinical studies have been conducted to evaluate the pharmacokinetics of tofersen in patients with renal or hepatic impairment. Tofersen is not expected to undergo metabolism by hepatic enzymes.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Studies to assess the carcinogenic potential of tofersen have not been conducted.
Mutagenesis
Tofersen was negative in in vitro (bacterial reverse mutation and mammalian cell chromosomal aberration) and in vivo (mouse micronucleus) assays.
Impairment of Fertility
In a study to assess effects on fertility and reproductive function, tofersen (0, 3, 10, 30 mg/kg) was administered every other day to male and female mice prior to and during mating and continuing in females to gestation day (GD) 7. Adverse effects on male reproductive organs (seminiferous tubular degeneration, seminiferous tubule dilatation, spermatid retention, apoptosis of epithelial cells, increased cellular debris in the testes, and hypospermia in the epididymis) were observed at the highest dose tested; however, there were no adverse effects on functional endpoints. Plasma exposure at the no-effect dose (10 mg/kg) for adverse effects on male reproductive organs was approximately 2 times that in humans at the recommended human dose of 100 mg.
14. Clinical Studies
The efficacy of QALSODY was assessed in a 28-week randomized, double-blind, placebocontrolled clinical study in patients 23 to 78 years of age with weakness attributable to ALS and a SOD1 mutation confirmed by a central laboratory (Study 1 Part C, NCT02623699). One hundred eight (108) patients were randomized 2:1 to receive treatment with either QALSODY 100 mg (n=72) or placebo (n=36) for 24 weeks (3 loading doses followed by 5 maintenance doses). Concomitant riluzole and/or edaravone use was permitted for patients.
The prespecified primary analysis population (n=60, modified intent to treat [mITT]) had a slow vital capacity (SVC) ≥65% of predicted value and met prognostic enrichment criteria for rapid disease progression, defined based on their pre-randomization ALS Functional Rating Scale–Revised (ALSFRS-R) decline slope and SOD1 mutation type.
The non-mITT population (n=48) had a slow vital capacity (SVC) ≥50% of predicted value and did not meet the enrichment criteria for rapid disease progression.
Baseline disease characteristics in the overall intent-to-treat (ITT) population (combined mITT and non-mITT population) were generally similar in patients treated with QALSODY and patients who received placebo, with slightly shorter time from symptom onset and higher plasma NfL at baseline in the QALSODY group. At baseline, 62% of patients were taking riluzole, and 8% of patients were taking edaravone. Mean baseline ALSFRS-R score was 36.9 (5.9) in the QALSODY treatment group and 37.3 (5.8) in the placebo group. Median time from symptom onset was 11.4 months in the QALSODY treatment group and 14.6 months in the placebo group.
The primary efficacy analysis was the change from baseline to Week 28 in the ALSFRS-R total score in the mITT population, analyzed using the joint rank test to account for mortality in conjunction with multiple imputation (MI) to account for missing data for withdrawals other than death. Patients treated with QALSODY experienced less decline from baseline in the ALSFRS-R compared to placebo, but the results were not statistically significant (QALSODY-placebo adjusted mean difference [95% CI]: 1.2 [-3.2, 5.5]). Other clinical secondary outcomes also did not reach statistical significance.
Secondary endpoints of change from baseline at Week 28 in plasma NfL and CSF SOD1 protein were nominally statistically significant (see Table 2). NfL reduction was consistently observed for all subgroups based on sex, disease duration since symptom onset, site of onset, and riluzole/edaravone use.
Table 2. Biomarker Results of QALSODY in Study 1 Part C at Week 28:
| Biomarker Endpoints | QALSODY | Placebo |
|---|---|---|
| Plasma NfL | ||
| ITT population | N=72 | N=36 |
| Adjusted geometric mean ratio to baseline | 0.45 | 1.12 |
| QALSODY to placebo difference in geometric mean ratio (95% CI) | 0.40 (0.33, 0.49) | |
| Nominal p-value (ANCOVA+MI) | <0.0001 | |
| mITT population | N=39 | N=21 |
| Adjusted geometric mean ratio to baseline | 0.40 | 1.20 |
| QALSODY to placebo difference in geometric mean ratio (95% CI) | 0.33 (0.25, 0.45) | |
| Nominal p-value (ANCOVA+MI) | <0.0001 | |
| CSF SOD1 Protein | ||
| ITT population | N=72 | N=36 |
| Adjusted geometric mean ratio to baseline | 0.65 | 0.98 |
| QALSODY to placebo difference in geometric mean ratio (95% CI) Nominal p-value (ANCOVA+MI) | 0.66 (0.57, 0.77) <0.0001 | |
| mITT population | N=39 | N=21 |
| Adjusted geometric mean ratio to baseline | 0.71 | 1.16 |
| QALSODY to placebo difference in geometric mean ratio (95% CI) | 0.62 (0.49, 0.78) | |
| Nominal p-value (ANCOVA+MI) | <0.0001 | |
Note 1: N is the number of patients with baseline value.
Note 2: MI was used for missing data. Model included treatment, use of riluzole or edaravone, relevant baseline score and postbaseline values (natural log transformed data). Separate models for mITT and nonmITT were used and combined for ITT analyses.
Note 3: Adjusted geometric mean ratios to baseline, treatment differences in adjusted geometric mean ratios to baseline and corresponding 95% CIs and nominal p-values were obtained from the ANCOVA model for change from baseline including treatment as a fixed effect and adjusting for the following covariates: baseline disease duration since symptom onset, relevant baseline score, and use of riluzole or edaravone. The analysis was based on natural log transformed data.
Figure 2. Plasma NfL Adjusted Geometric Mean Ratio to Baseline Values in Study 1 Part C by Study Week for the ITT Population:
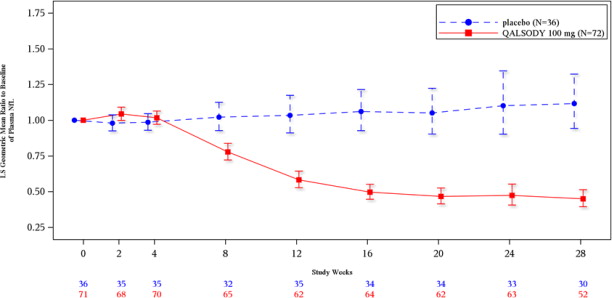
After completion of Study 1, patients had the option to enroll in an open-label extension study. At an interim analysis at 52 weeks, reductions in NfL were seen in patients previously receiving placebo who initiated QALSODY in the open-label extension study, similar to the reductions seen in patients treated with QALSODY in Study 1. Earlier initiation of QALSODY compared to placebo/delayed initiation of QALSODY was associated with trends for reduction in decline on ALSFRS-R, SVC percent-predicted, and hand-held dynamometry (HHD) megascore that were not statistically significant. Through all open-label follow-up at the time of the interim analysis, earlier initiation of QALSODY was also associated with a trend towards reduction of the risk of death or permanent ventilation, although it was not statistically significant. These exploratory analyses should be interpreted with caution given the limitations of data collected outside of a controlled study, which may be subject to confounding.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.