TEVIMBRA Concentrate for solution for infusion Ref.[51271] Active ingredients: Tislelizumab
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: BeOne Medicines Ireland Limited, 10 Earlsfort Terrace, Dublin 2, D02 T380, Ireland, Tel. +353 1 566 7660, E-mail: beone.ireland@beonemed.com
5.1. Pharmacodynamic properties
Pharmacotherapeutic group:Antineoplastic agents, monoclonal antibodies and antibody drug conjugates
ATC code: L01FF09
Mechanism of action
Tislelizumab is a humanised immunoglobulin G4 (IgG4) variant monoclonal antibody against PD-1, binding to the extracellular domain of human PD-1. It competitively blocks the binding of both PD-L1 and PD-L2, inhibiting PD-1-mediated negative signalling and enhancing the functional activity in T-cells in in vitro cell-based assays.
Clinical efficacy and safety
Non-small cell lung cancer
Neoadjuvant and adjuvant treatment of resectable NSCLC: BGB-A317-315
BGB-A317-315 was a phase 3 randomised, placebo-controlled, double-blind study to compare the efficacy and safety of neoadjuvant treatment with tislelizumab plus platinum-based doublet chemotherapy followed by adjuvant tislelizumab versus neoadjuvant treatment with placebo plus platinum-based doublet chemotherapy followed by adjuvant placebo in patients with resectable Stage II or IIIA NSCLC.
The study included patients who had histologically confirmed Stage II or IIIA (AJCC 8th) NSCLC with ECOG PS of 0 or 1 and no known EGFR mutations or ALK gene translocations and confirmed eligibility for R0 resection with curative intent. Patients with Stage IIIB were not included in the study.
The following selection criteria define patients with high risk of recurrence who are included in the therapeutic indication and are reflective of the patient population with Stage II – IIIA according to the 8th edition AJCC staging system:
- Tumour size >4 cm; or tumours of any size that are either accompanied by N1 or N2 status;
- Tumours that invade thoracic structures (directly invade the visceral pleura, parietal pleura, chest wall, main bronchus, phrenic nerve, mediastinal pleura, parietal pericardium);
- Tumours >4 cm that cause obstructive atelectasis that extends to the hilar region, involving part or all of the lung or involve a mainstem bronchus regardless of distance to the carina, or invades visceral pleura (PL1 or PL2) for N0 status;
- Tumours with separate nodule(s) in the same lobe as the primary lung cancer.
A total of 453 patients were randomised (1:1) to receive:
- Tislelizumab arm: neoadjuvant tislelizumab 200 mg on Day 1 in combination with either cisplatin 75 mg/m² or carboplatin AUC of 5 mg/mL/min and pemetrexed 500 mg/m² or paclitaxel 175 mg/m² on Day 1 of each 21-day cycle for 3 to 4 cycles. Following surgery, adjuvant tislelizumab 400 mg was administered every 6 weeks for up to 8 cycles.
- Placebo arm: neoadjuvant placebo on Day 1 in combination with either cisplatin 75 mg/m² or carboplatin AUC of 5 mg/mL/min and pemetrexed 500 mg/m² or paclitaxel 175 mg/m² on Day 1 of each 21-day cycle for 3 to 4 cycles. Following surgery, adjuvant placebo was administered every 6 weeks for up to 8 cycles.
Patients with non-squamous histology received pemetrexed while patients with squamous histology received paclitaxel, whereby the choice of cisplatin or carboplatin was decided by the investigators for all patients. If indicated, patients received postoperative adjuvant radiation therapy prior to adjuvant tislelizumab or placebo. Administration of tislelizumab and chemotherapy continued until treatment completion, disease progression, unacceptable AE, death, or patient and/or investigator's decision to discontinue study treatment.
The dual primary endpoints were event-free survival (EFS) by blinded independent central review (BICR) and major pathological response (MPR) rate by blinded independent pathological review (BIPR). The secondary efficacy endpoints included pathological complete response (pCR) rate by BIPR, and overall survival (OS).
Demographics and baseline characteristics were generally balanced between the 2 treatment arms. The baseline characteristics for all 453 randomised patients were: median age of 62 years (range 30 to 80 years); 40% of patients were ≥65 years of age; 3.3% of patients were ≥75 years of age; 90.5% of patients were male; 100% Asian (all enrolled in China), 65.3% had an ECOG PS score of 0; 84.5% were current or former smokers; 78.1% had diagnosed squamous histology; 58.5% had stage IIIA disease; 57.8% had PD-L1 expression ≥1%.
There were 84.1% of patients in the tislelizumab in combination with platinum-containing chemotherapy arm who had definitive surgery compared to 76.2% of patients in the platinum-containing chemotherapy arm.
The study demonstrated statistically significant improvement in MPR, EFS, pCR and OS for patients randomised to tislelizumab arm compared with placebo arm.
At a prespecified interim analysis of EFS (data cut-off date 21-Aug-2023), the EFS HR was 0.56 (95% CI: 0.40, 0.79; 1-sided p-value of 0.0003) and the median OS follow-up times by reverse Kaplan-Meier methodology were 24.6 months in the tislelizumab arm and 22.7 months in the placebo arm.
Table 3, Figure 1 and Figure 2 summarise the efficacy results.
At a pre-specified final analysis (data cut-off date 07-Mar-2025), the median OS follow-up times by reverse Kaplan-Meier methodology were 43.3 months (95% CI: 41.2, 44.6) in the tislelizumab arm and 41.6 months (95% CI: 39.9, 43.8) in the placebo arm.
Table 3. Efficacy results in BGB-A317-3151:
| Tislelizumab arm (N=226) | Placebo arm (N=227) | |
|---|---|---|
| Event-Free Survival | ||
| Events, n (%) | 72 (31.9) | 98 (43.2) |
| Median (months) (95% CI) | NR (50.3, NE) | 30.6 (16.6, 45.3) |
| HR (95% CI)a | 0.58 (0.43, 0.79) | |
| Major Pathological Response | ||
| n (%) | 127 (56.2) | 34 (15) |
| 95% CIc | (49.5, 62.8) | (10.6, 20.3) |
| Difference, % (95% CI)d | 41.1 (33.2, 49.1) | |
| p-valuee | <0.0001 | |
| Overall Survival | ||
| Deaths, n (%) | 52 (23.0) | 70 (30.8) |
| Median (months) (95% CI) | NR (NE, NE) | NR (NE, NE) |
| HR (95% CI)a | 0.65 (0.45, 0.93) | |
| p-valueb | 0.0093 | |
CI = confidence interval; HR = hazard ratio; NE = not estimable; NR = not reached
Patients without surgery or pathological results were considered as non-responders.
1 The prespecified final analysis of MPR was based on the data with cut-off date of 20-Feb-2023 and the prespecified final analysis of EFS and OS was based on the data with cut-off date of 07-Mar-2025.
a Hazard ratio and 95% CIs were estimated using a stratified Cox regression model stratified by histology, disease stage and PD-L1 expression from interactive response technology (IRT).
b The p-value was calculated using a log-rank test stratified by histology, disease stage and PD-L1 expression from IRT.
c The 95% CI was estimated using the Clopper-Pearson method.
d Mantel-Haenszel common risk difference was estimated along with its 95% CIs constructed by a normal approximation and Sato's variance estimator stratified by histology, disease stage and PD-L1 expression from IRT.
e The p-value was obtained using the Cochran-Mantel-Haenszel method stratified by histology, disease stage and PD-L1 expression from IRT.
Figure 1. Kaplan-Meier Plot for Event-Free Survival in BGB-A317-315:

Figure 2. Kaplan-Meier Plot for Overall Survival in BGB-A317-315:

A subgroup analysis was performed in study BGB-A317-315 in patients who had PD-L1 ≥1% (tislelizumab arm [n=130; 58%] vs. placebo arm [n=132; 58%]) and PD-L1 <1% (which excludes not evaluable/indeterminate) (tislelizumab arm [n=89; 39%] vs. placebo arm [n=84; 37%]). The EFS HR was 0.53 (95% CI: 0.35, 0.79) in patients with PD-L1 ≥1% and 0.70 (95% CI: 0.43, 1.14) in patients with PD-L1 <1%. The OS HR was 0.61 (95% CI: 0.38, 0.98) in patients with PD-L1 ≥1% and 0.91 (95% CI: 0.50, 1.64) in patients with PD-L1 <1%.
First-line treatment of non-squamous NSCLC: BGB-A317-304
BGB-A317-304 was a randomised, open-label, multicentre phase III study to investigate the efficacy and safety of tislelizumab in combination with platinum-pemetrexed versus platinum-pemetrexed alone as first-line treatment for chemotherapy-naïve patients with locally advanced non-squamous NSCLC who were not candidates for surgical resection or platinum-based chemoradiation, or metastatic non-squamous NSCLC.
The study excluded patients with active brain or leptomeningeal metastases, known EGFR mutations or ALK translocations sensitive to available targeted inhibitor therapy, active autoimmune disease, or any condition requiring systemic treatment with either corticosteroids (>10 mg daily of prednisone or equivalent) or other immunosuppressants.
A total of 334 patients were randomised (2:1) to receive tislelizumab 200 mg combined with pemetrexed 500 mg/m² and carboplatin AUC 5 mg/ml/min or cisplatin 75 mg/m² (T+PP arm, N=223) or pemetrexed 500 mg/m² and carboplatin AUC 5 mg/ml/min or cisplatin 75 mg/m² (PP arm, N=111). The choice of platinum (cisplatin or carboplatin) was at the investigator's discretion.
The treatment was administered on a 3-week cycle. After the administration of 4, 5 or 6 cycles of chemotherapy or tislelizumab combined with chemotherapy at the investigator's discretion, patients in the T+PP arm received tislelizumab 200 mg combined with pemetrexed 500 mg/m² on a 3-week cycle until disease progression or unacceptable toxicity; patients in the PP arm received pemetrexed 500 mg/m² alone until disease progression or unacceptable toxicity, and those with disease progression confirmed by Independent Review Committee (IRC) were given the option to cross over to receive tislelizumab monotherapy on a 3-week cycle.
Randomisation was stratified by PD-L1 expression in tumour cells (TC) (<1% versus 1% to 49% versus ≥50%) and disease stage (IIIB versus IV), as classified according to American Joint Committee on Cancer (AJCC), 7th edition of Cancer Staging Manual. PD-L1 expression was evaluated at a central laboratory using the Ventana PD-L1 (SP263) assay that identified PD-L1 staining on tumour cells. Tumour assessments were conducted every 6 weeks for the first 6 months, then every 9 weeks for the second 6 months, then every 12 weeks.
The baseline characteristics for patients in study BGB-A317-304 were: median age 61 years (range: 25 to 75), 29% age 65 years or older; 74% male; 100% Asian (all enrolled in China); 23.4% with ECOG PS of 0 and 76.6% with ECOG PS of 1; 18.3% with disease stage IIIB; 26.6% with unknown status for ALK rearrangement and 73.4% with negative ALK rearrangement; 36.2% never-smokers; 5.4% with brain metastases. The characteristics of age, sex, ECOG PS, stage, smoking status, PD-L1 TC score and prior anticancer treatments were balanced between the treatment arms.
The primary efficacy endpoint was progression-free survival (PFS) per RECIST v1.1 by IRC in the intent-to-treat (ITT) analysis. The secondary efficacy endpoints included overall survival (OS), objective response rate (ORR) and duration of response (DoR) per IRC and per investigator.
The study met its primary endpoint at the interim analysis (data cut-off date of 23-Jan-2020), showing a statistically significant improvement in PFS with T+PP compared with PP. The stratified hazard ratio was 0.65 (95% CI: 0.47, 0.91; p=0.0054) with a median PFS of 9.7 months with T+PP and 7.6 months with PP. The median OS follow-up times by reverse Kaplan-Meier methodology were 9.9 months in the T+PP arm and 9.7 months in the PP arm.
The efficacy results of the final analysis (data cut-off date of 26-Oct-2020) were consistent with those of the interim analysis. At the final analysis, the median OS follow-up times by reverse Kaplan-Meier methodology were 18.4 months in the T+PP arm and 18.0 months in the PP arm.
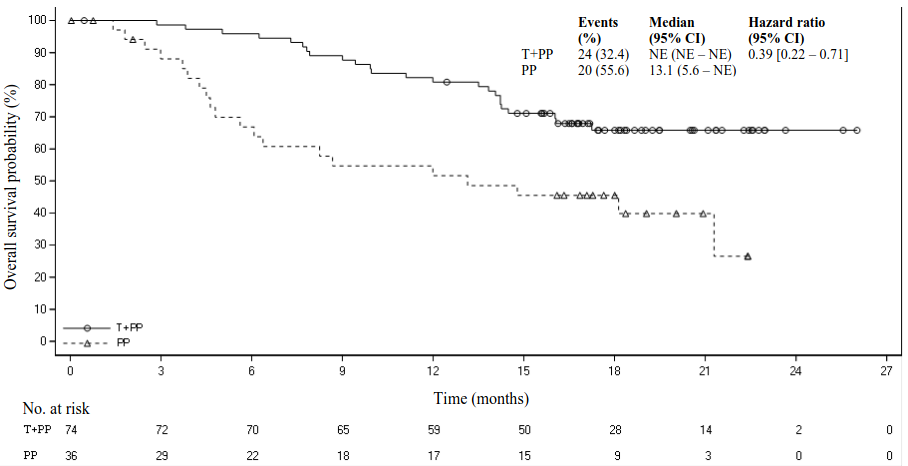
Amongst the 334 patients in study BGB-A317-304, 110 (33%) patients had tumour cell PD-L1 expression ≥50%. Of these, 74 patients were in the tislelizumab plus chemotherapy group and 36 patients were in the placebo plus chemotherapy group. Efficacy results of the patients with tumour cell PD-L1 expression ≥50% from the final analysis are shown in Table 4 and the Kaplan-Meier curve for PFS and OS is presented in Figures 3 and 4, respectively.
Table 4. Efficacy results in BGB-A317-304 in patients with PD-L1 expression ≥50%:
| Endpoint | Tislelizumab + Pemetrexed + Platinum (n=74) | Pemetrexed + Platinum (n=36) |
|---|---|---|
| PFS | ||
| Events, n (%) | 33 (44.6) | 22 (61.1) |
| Median PFS (months) (95% CI) | 14.6 (11.5, NE) | 4.6 (3.5, 9.7) |
| Stratified hazard ratioa (95% CI) | 0.31 (0.18, 0.55) | |
| OS | ||
| Deaths, n (%) | 24 (32.4) | 20 (55.6) |
| Median OS (months) (95% CI) | NE (NE, NE) | 13.1 (5.6, NE) |
| Stratified hazard ratioa (95% CI) | 0.39 (0.22, 0.71) | |
| Best overall response, n (%)b | ||
| ORRb, n (%) | 52 (70.3) | 11 (30.6) |
| 95% CIc | (58.5, 80.3) | (16.3, 48.1) |
| DoRb | ||
| Median DoR (months) (95% CI) | NE (13.2, NE) | 8.5 (3.3 NE) |
PFS = progression-free survival; CI = confidence interval; OS = overall survival; ORR = objective response rate; DoR = duration of response; NE = not estimable.
Medians were estimated by Kaplan-Meier method with 95% CIs estimated using the method of Brookmeyer and Crowley.
a Hazard ratio was estimated from stratified Cox model with pemetrexed+platinum group as reference group and stratified by disease stage (IIIB versus IV).
b PFS was based on IRC assessment, and ORR/DoR was based on the confirmed response by IRC.
c 95% CI was calculated using Clopper-Pearson method.
Figure 3. Kaplan‑Meier plot of PFS in BGB‑A317‑304 in patients with PD-L1 ≥50%:

Figure 4. Kaplan-Meier plot of OS in BGB-A317-304 in patients with PD-L1 ≥50%:

First-line treatment of non-squamous NSCLC: BGB-A317-304
BGB-A317-304 was a randomised, open-label, multicentre phase III study to investigate the efficacy and safety of tislelizumab in combination with platinum-pemetrexed versus platinum-pemetrexed alone as first-line treatment for chemotherapy-naïve patients with locally advanced non-squamous NSCLC who were not candidates for surgical resection or platinum-based chemoradiation, or metastatic non-squamous NSCLC.
The study excluded patients with active brain or leptomeningeal metastases, known EGFR mutations or ALK translocations sensitive to available targeted inhibitor therapy, active autoimmune disease, or any condition requiring systemic treatment with either corticosteroids (>10 mg daily of prednisone or equivalent) or other immunosuppressants.
A total of 334 patients were randomised (2:1) to receive tislelizumab 200 mg combined with pemetrexed 500 mg/m² and carboplatin AUC 5 mg/ml/min or cisplatin 75 mg/m² (T+PP arm, N=223) or pemetrexed 500 mg/m² and carboplatin AUC 5 mg/ml/min or cisplatin 75 mg/m² (PP arm, N=111). The choice of platinum (cisplatin or carboplatin) was at the investigator's discretion.
The treatment was administered on a 3-week cycle. After the administration of 4, 5 or 6 cycles of chemotherapy or tislelizumab combined with chemotherapy at the investigator's discretion, patients in the T+PP arm received tislelizumab 200 mg combined with pemetrexed 500 mg/m² on a 3-week cycle until disease progression or unacceptable toxicity; patients in the PP arm received pemetrexed 500 mg/m² alone until disease progression or unacceptable toxicity, and those with disease progression confirmed by Independent Review Committee (IRC) were given the option to cross over to receive tislelizumab monotherapy on a 3-week cycle.
Randomisation was stratified by PD-L1 expression in tumour cells (TC) (<1% versus 1% to 49% versus ≥50%) and disease stage (IIIB versus IV), as classified according to American Joint Committee on Cancer (AJCC), 7th edition of Cancer Staging Manual. PD-L1 expression was evaluated at a central laboratory using the Ventana PD-L1 (SP263) assay that identified PD-L1 staining on tumour cells. Tumour assessments were conducted every 6 weeks for the first 6 months, then every 9 weeks for the second 6 months, then every 12 weeks.
The baseline characteristics for patients in study BGB-A317-304 were: median age 61 years (range: 25 to 75), 29% age 65 years or older; 74% male; 100% Asian (all enrolled in China); 23.4% with ECOG PS of 0 and 76.6% with ECOG PS of 1; 18.3% with disease stage IIIB; 26.6% with unknown status for ALK rearrangement and 73.4% with negative ALK rearrangement; 36.2% never-smokers; 5.4% with brain metastases. The characteristics of age, sex, ECOG PS, stage, smoking status, PD-L1 TC score and prior anticancer treatments were balanced between the treatment arms.
The primary efficacy endpoint was progression-free survival (PFS) per RECIST v1.1 by IRC in the intent-to-treat (ITT) analysis. The secondary efficacy endpoints included overall survival (OS), objective response rate (ORR) and duration of response (DoR) per IRC and per investigator.
The study met its primary endpoint at the interim analysis (data cut-off date of 23-Jan-2020), showing a statistically significant improvement in PFS with T+PP compared with PP. The stratified hazard ratio was 0.65 (95% CI: 0.47, 0.91; p=0.0054) with a median PFS of 9.7 months with T+PP and 7.6 months with PP. The median OS follow-up times by reverse Kaplan-Meier methodology were 9.9 months in the T+PP arm and 9.7 months in the PP arm.
The efficacy results of the final analysis (data cut-off date of 26-Oct-2020) were consistent with those of the interim analysis. At the final analysis, the median OS follow-up times by reverse Kaplan-Meier methodology were 18.4 months in the T+PP arm and 18.0 months in the PP arm.
Amongst the 334 patients in study BGB-A317-304, 110 (33%) patients had tumour cell PD-L1 expression ≥50%. Of these, 74 patients were in the tislelizumab plus chemotherapy group and 36 patients were in the placebo plus chemotherapy group. Efficacy results of the patients with tumour cell PD-L1 expression ≥50% from the final analysis are shown in Table 3 and the Kaplan-Meier curve for PFS and OS is presented in Figures 1 and 2, respectively.
Table 3. Efficacy results in BGB-A317-304 in patients with PD-L1 expression ≥50%:
| Endpoint | Tislelizumab + Pemetrexed + Platinum (N=74) | Pemetrexed + Platinum (N=36) |
|---|---|---|
| PFS | ||
| Events, n (%) | 33 (44.6) | 22 (61.1) |
| Median PFS (months) (95% CI) | 14.6 (11.5, NE) | 4.6 (3.5, 9.7) |
| Stratified hazard ratioa (95% CI) | 0.31 (0.18, 0.55) | |
| OS | ||
| Deaths, n (%) | 24 (32.4) | 20 (55.6) |
| Median OS (months) (95% CI) | NE (NE, NE) | 13.1 (5.6, NE) |
| Stratified hazard ratioa (95% CI) | 0.39 (0.22, 0.71) | |
| Best overall response, n (%) b | ||
| ORRb, n (%) | 52 (70.3) | 11 (30.6) |
| 95% CIc | (58.5, 80.3) | (16.3, 48.1) |
| DoRb | ||
| Median DoR (months) (95% CI) | NE (13.2, NE) | 8.5 (3.3 NE) |
PFS = progression-free survival; CI = confidence interval; OS = overall survival; ORR = objective response rate; DoR = duration of response; NE = not estimable.
Medians were estimated by Kaplan-Meier method with 95% CIs estimated using the method of Brookmeyer and Crowley.
a Hazard ratio was estimated from stratified Cox model with pemetrexed+platinum group as reference group and stratified by disease stage (IIIB versus IV).
b PFS was based on IRC assessment, and ORR/DoR was based on the confirmed response by IRC.
c 95% CI was calculated using Clopper-Pearson method.
Figure 1. Kaplan‑Meier plot of PFS in BGB‑A317‑304 in patients with PD-L1 ≥50%:

Figure 2. Kaplan-Meier plot of OS in BGB-A317-304 in patients with PD-L1 ≥50%:
First-line treatment of squamous NSCLC: BGB-A317-307
BGB-A317-307 was a randomised, open-label, multicentre phase III study to compare the efficacy and safety of tislelizumab in combination with paclitaxel plus carboplatin or nab-paclitaxel plus carboplatin with that of paclitaxel plus carboplatin alone as first-line treatment for chemotherapy-naïve patients with locally advanced squamous NSCLC who were not candidates for surgical resection or platinum-based chemoradiation or metastatic squamous NSCLC.
The study excluded patients with active brain or leptomeningeal metastases, known EGFR mutations or ALK translocations sensitive to available targeted inhibitor therapy, active autoimmune disease, or any condition requiring systemic treatment with either corticosteroids (>10 mg daily of prednisone or equivalent) or other immunosuppressive treatments.
A total of 360 patients were randomised (1:1:1) to receive tislelizumab 200 mg combined with paclitaxel 175 mg/m² and carboplatin AUC 5 mg/ml/min (T+PC arm, N=120), or tislelizumab 200 mg combined with nab-paclitaxel 100 mg/m² and carboplatin AUC 5 mg/ml/min (T+nPC arm, N=119), or paclitaxel 175 mg/m² and carboplatin AUC 5 mg/ml/min (PC arm, N=121).
The treatment was administered on a 3-week cycle, until the patient completed administration of 4 to 6 cycles of chemotherapy or tislelizumab combined with chemotherapy at the investigator's discretion. Patients in the T+nPC and T+PC arms received tislelizumab until disease progression or unacceptable toxicity. Patients in the PC arm with disease progression were given the option to cross over to receive tislelizumab monotherapy on a 3-week cycle.
Randomisation was stratified by PD-L1 expression in tumour cells (TC) (<1% versus 1% to 49% versus ≥50%) and tumour staging (IIIB versus IV), as classified according to American Joint Committee on Cancer (AJCC), 7th edition of Cancer Staging Manual. PD-L1 expression was evaluated at a central laboratory using the Ventana PD-L1(SP263) assay that identified PD-L1 staining on tumour cells. Tumour assessments were conducted every 6 weeks for the first 6 months, then every 9 weeks for the remainder of the first year, then every 12 weeks until disease progression.
The baseline characteristics for the study population were: median age 62.0 years (range: 34 to 74), 35.3% age 65 years or older; 91.7% male; 100% Asian (all enrolled in China), 23.6% with ECOG PS of 0 and 76.4% with ECOG PS of 1; 33.9% diagnosed with stage IIIB and 66.1% with stage IV at baseline; 16.4% never-smokers; 38.3% with PD-L1 TC score <1%, 25.3% with PD-L1 TC score ≥1% and ≤49%, 34.7% with PD-L1 TC score ≥50%. The characteristics of age, sex, ECOG PS, stage, smoking status, PD-L1 TC score and prior anticancer treatments were balanced between the treatment arms.
The primary efficacy endpoint was progression-free survival (PFS) as assessed by IRC per RECIST v1.1 in the ITT analysis which was to be tested sequentially in arms T+PC versus PC and arms T+nPC versus PC. The secondary efficacy endpoints included overall survival (OS), objective response rate (ORR) and duration of response (DoR) per IRC and per investigator.
The study met its primary endpoint at the interim analysis (data cut-off date of 06-Dec-2019), showing statistically significant improvements in PFS with tislelizumab in combination with paclitaxel and carboplatin (T+PC arm) and tislelizumab in combination with nab-paclitaxel and carboplatin (T+nPC arm) compared with paclitaxel and carboplatin alone (PC arm). The stratified HR (T+PC arm versus PC arm) was 0.48 (95% CI: 0.34, 0.69; p<0.0001). The stratified HR (T+nPC arm versus PC arm) was 0.45 (95% CI: 0.32, 0.64; p<0.0001). Median PFS was 7.6 months in the T+PC arm, 7.6 months in the T+nPC arm and 5.4 months in the PC arm. The median OS follow-up times by reverse Kaplan-Meier methodology were 8.8 months in the T+PC arm, 8.8 months in the T+nPC arm, and 8 months in the PC arm.
The final analysis (data cut-off date of 30-Sep-2020) showed the consistent results from the interim analysis. At the final analysis, the median OS follow-up times by reverse Kaplan-Meier methodology were 18.8 months in the T+PC arm, 18.9 months in the T+nPC arm, and 18.1 months in the PC arm.
Efficacy results for the final analysis are shown in Table 5, Figure 5 and Figure 6.
Table 5. Efficacy results in BGB-A317-307:
| Endpoint | Tislelizumab + Paclitaxel + Carboplatin (N=120) | Tislelizumab + nab-Paclitaxel + Carboplatin (N=119) | Paclitaxel + Carboplatin (N=121) |
|---|---|---|---|
| PFS | |||
| Events, n (%) | 80 (66.7) | 79 (66.4) | 86 (71.1) |
| Median PFS (months) (95% CI) | 7.7 (6.7, 10.4) | 9.6 (7.4, 10.8) | 5.5 (4.2, 5.6) |
| Stratified hazard ratioa (95% CI) | 0.45 (0.33, 0.62) | 0.43 (0.31, 0.60) | - |
| OS | |||
| Deaths, n (%) | 48 (40.0) | 47 (39.5) | 52 (43.0) |
| Median OS (months) (95% CI) | 22.8 (19.1, NE) | NE (18.6, NE) | 20.2 (16.0, NE) |
| Stratified hazard ratio (95% CI) | 0.68 (0.45, 1.01) | 0.752 (0.50, 1.12) | - |
| ORRb | |||
| ORR, n (%) | 74 (61.7) | 74 (62.2) | 45 (37.2) |
| 95% CI | (52.4, 70.4) | (52.8, 70.9) | (28.6, 46.4) |
| DoRb | |||
| Median DoR (months) (95% CI) | 13.2 (7.85, 18.79) | 10.4 (8.34, 17.15) | 4.8 (4.04, 5.72) |
PFS = progression-free survival; CI = confidence interval; OS = overall survival; ORR = objective response rate; DoR = duration of response; NE = not estimable.
a Stratified by stratification factors: disease stage (IIIB versus IV) and PD‑L1 expression in tumour cell (≥50% TC versus 1% to 49% TC versus <1% TC).
b PFS was based on IRC assessment, and ORR/DoR was based on the confirmed response by IRC.
Figure 5. Kaplan-Meier plot of PFS in BGB-A317-307 by IRC:
T+PC arm versus T+nPC arm versus PC arm
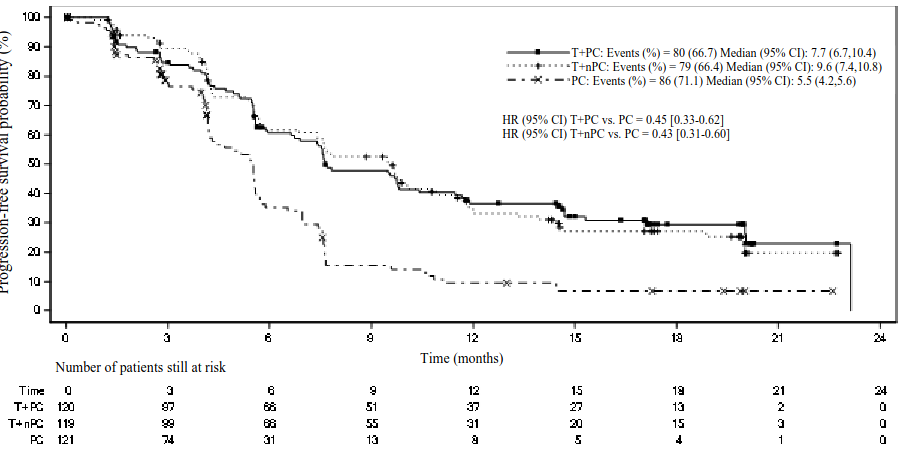
CI = Confidence interval; T+PC = tislelizumab+paclitaxel+carboplatin; T+nPC = tislelizumab+nab‑paclitaxel+carboplatin; PC = paclitaxel+carboplatin.
Figure 6. Kaplan-Meier plot of OS in BGB-A317-307:
T+PC arm versus T+nPC arm versus PC arm
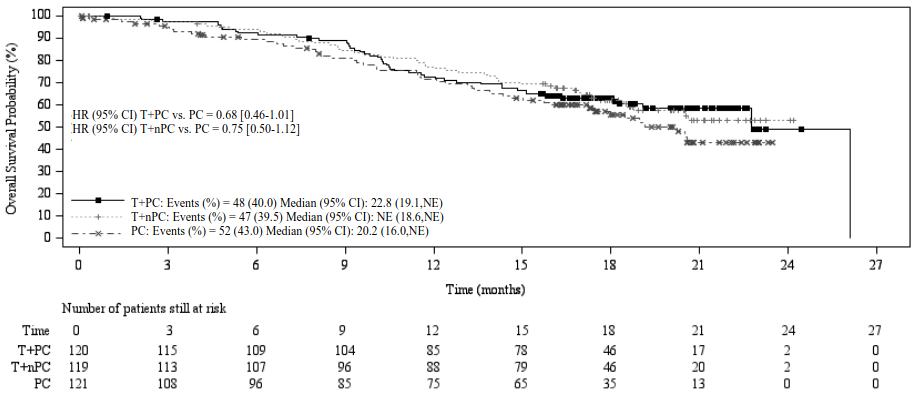
CI = Confidence interval; T+PC = tislelizumab+paclitaxel+carboplatin; T+nPC = tislelizumab+nab-paclitaxel+carboplatin; PC = paclitaxel+carboplatin; NE = not estimable.
Subgroup analyses demonstrated consistent PFS treatment effect across major demographic and prognostic subgroups, including PD-L1 expression <1%, 1 to 49% and ≥50% and disease stages IIIB and IV:
- for T+PC, with PFS HR of 0.57 (95% CI, HR = 0.34, 0.94) for PD-L1 <1%, 0.40 (95% CI, HR = 0.21, 0.76) for 1 to 49% and 0.44 (95% CI, HR = 0.26, 0.75) for ≥50%
- for T+nPC, with PFS HR of 0.65 (95% CI, HR = 0.40, 1.06) for PD-L1 <1%, 0.40 (95% CI, HR = 0.22, 0.74) for 1 to 49% and 0.33 (95% CI, HR = 0.18, 0.59) for ≥50%
Previously treated NSCLC: BGB-A317-303
BGB-A317-303 was a randomised, open-label, multicentre phase III study to investigate the efficacy and safety of tislelizumab compared with docetaxel in patients with locally advanced or metastatic NSCLC (squamous or non-squamous), who had experienced disease progression on or after a prior platinum-based regimen.
The study excluded patients with known EGFR mutation or ALK rearrangement, prior PD-(L)1 inhibitor or CTLA-4 inhibitor treatment, active autoimmune disease, or any condition requiring systemic treatment with either corticosteroids (>10 mg daily of prednisone or equivalent) or other immunosuppressive treatments.
A total of 805 patients were randomised (2:1) ratio to receive tislelizumab 200 mg intravenously every 3 weeks (N=535) or docetaxel 75 mg/m² intravenously every 3 weeks (N=270). Randomisation was stratified by histology (squamous versus non-squamous), lines of therapy (second-versus third-line), and PD-L1 expression in tumour cells (TC) (≥25% versus <25%). Administration of docetaxel and tislelizumab continued until disease progression, as assessed by investigator per RECIST v1.1, or unacceptable toxicity. PD-L1 expression was evaluated at a central laboratory using the Ventana PD-L1 (SP263) assay that identified PD-L1 staining on tumour cells. Tumour assessments were conducted every 9 weeks for 52 weeks after randomisation and continued every 12 weeks thereafter. Survival status was followed every 3 months after discontinuation of the study treatment.
The baseline characteristics for the study population were: median age 61 years (range: 28 to 88), 32.4% age 65 years or older, 3.2% age 75 years or older; 77.3% male; 17.0% White and 79.9% Asian; 20.6% with ECOG PS of 0 and 79.4% with ECOG PS of 1; 85.5% with metastatic disease; 30.3% never-smokers; 46.0% with squamous and 54.0% non-squamous histology; 65.8% with wild-type and 34% with unknown EGFR status; 46.1% with wild-type and 53.9% with unknown ALK status; 7.1% with previously treated brain metastases.
57.0% of the patients had a PD-L1 TC score <25% and 42.5% had a PD-L1 TC score ≥25%. All patients had received prior therapy with a platinum-doublet regimen: 84.7% patients received one prior therapy, 15.3% had received two prior therapies.
The dual-primary efficacy endpoints were OS in the ITT and PD-L1 TC score ≥25% analysis sets. Additional efficacy endpoints included investigator-assessed PFS, ORR and DoR.
BGB-A317-303 met both dual-primary endpoints of OS in the ITT analysis and PD-L1 ≥25% analysis sets. At the prespecified interim analysis (data cut-off date 10-Aug-2020), a statistically significant improvement in OS was observed in the ITT population. Results favoured the tislelizumab arm (HR=0.64; 95% CI: 0.53, 0.78; p<0.0001). Median OS was 17.2 months for the tislelizumab arm and 11.9 months for the docetaxel arm. The median follow-up times by reverse Kaplan-Meier methodology were 19.5 months in the tislelizumab arm and 17.0 months in the docetaxel arm.At the final analysis (data cutoff date 15-Jul-2021), a statistically significant improvement in OS was observed in the PD-L1 ≥25% analysis set favouring the tislelizumab arm (stratified HR=0.53; 95% CI: 0.41, 0.70; p<0.0001) with median OS being 19.3 months for the tislelizumab arm and 11.5 months for the docetaxel arm. The median follow-up time by reverse Kaplan-Meier methodology at the final analysis were 31.1 months in the tislelizumab arm and 27.9 months in the docetaxel arm.
The final analysis (data cut-off date 15-Jul-2021) showed consistent efficacy results in the ITT population compared to the interim analysis.
Table 6 and Figure 7 summarise the efficacy results for BGB-A317-303 (ITT analysis set) at the final analysis.
Table 6. Efficacy results in BGB-A317-303:
| Endpoint | Tislelizumab (N=535) | Docetaxel (N=270) |
|---|---|---|
| OS | ||
| Deaths, n (%) | 365 (68.2) | 206 (76.3) |
| Median OS (months) (95% CI) | 16.9 (15.24, 19.09) | 11.9 (9.63, 13.54) |
| Hazard ratio (95% CI)a,b | 0.66 (0.56, 0.79) | |
| PFS | ||
| Events, n (%) | 451 (84.3) | 208 (77.0) |
| Median PFS (months) (95% CI) | 4.2 (3.88, 5.52) | 2.6 (2.17, 3.78) |
| Hazard ratioa (95% CI) | 0.63 (0.53, 0.75) | |
| ORR (%) (95% CI)c | 20.9 (17.56, 24.63) | 3.7 (1.79, 6.71) |
| DoRc | ||
| Median DoR (months) (95% CI) | 14.7 (10.55, 21.78) | 6.2 (4.11, 8.31) |
OS = overall survival; CI = confidence interval; PFS = progression-free survival; ORR = objective response rate; DoR = duration of response.
Medians were estimated by Kaplan-Meier method with 95% CIs estimated using the method of Brookmeyer and Crowley.
a Hazard ratio was estimated from stratified Cox model with docetaxel group as reference group.
b Stratified by stratification factors: histology (squamous versus non‑squamous), lines of therapy (second versus third), and PD-L1 expression in tumour cells (≥25% PD-L1 score versus <25% PD-L1 score).
c Confirmed by investigator.
Figure 7. Kaplan-Meier plot of OS in BGB-A317-303 (ITT Analysis Set):
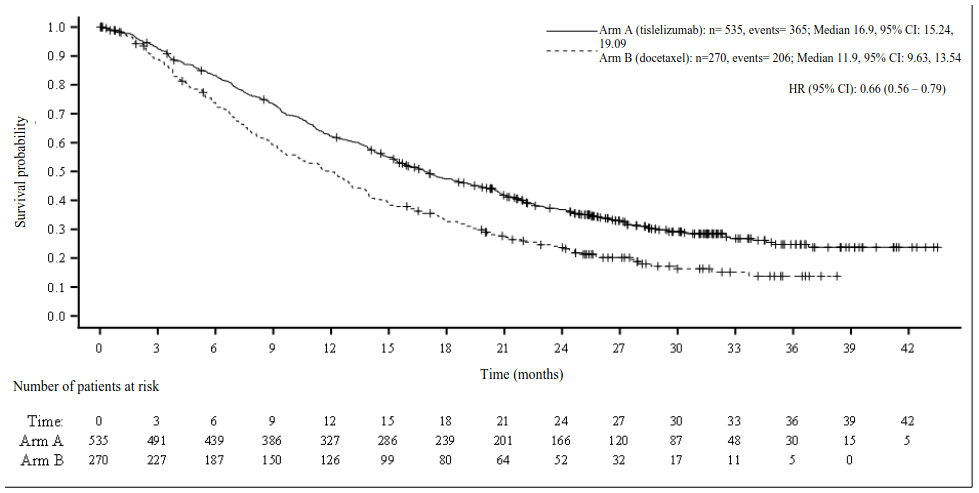
Prespecified subgroup analyses demonstrated a consistent OS treatment effect in favour of tislelizumab across major demographic and prognostic subgroups.
Table 7 summarises efficacy results of OS by tumour PD-L1 (<25% TC, ≥25% TC) expression in prespecified subgroup analyses.
Table 7. Efficacy results of OS by tumour PD-L1 expression (<25% TC, ≥25% TC) in BGB-A317-303:
| Tislelizumab arm | Docetaxel arm | |
|---|---|---|
| N=535 | N=270 | |
| PD‑L1 expression in tumour cells <25%, n | 307 | 152 |
| Events, n (%) | 223 (72.6) | 117 (77.0) |
| Median OS (months) (95% CI) | 15.2 (13.4, 17.6) | 12.3 (9.3, 14.3) |
| Hazard ratioa (95% CI) | 0.79 (0.64, 0.99) | |
| PD‑L1 expression in tumour cells ≥25%, n | 227 | 115 |
| Events, n (%) | 141 (62.1) | 86 (74.8) |
| Median OS (months) (95% CI) | 19.3 (16.5, 22.6) | 11.5 (8.2, 13.5) |
| Hazard ratioa (95% CI) | 0.54 (0.41, 0.71) | |
a Hazard ratio and its 95% CI were estimated from unstratified Cox model.
Small cell lung cancer
First-line treatment of extensive-stage SCLC: BGB-A317-312
BGB-A317-312 was a randomised, double-blind, multicentre phase III study to compare the efficacy and safety of tislelizumab in combination with cisplatin or carboplatin plus etoposide versus placebo in combination with cisplatin or carboplatin plus etoposide as first-line treatment in patients with extensive-stage small cell lung cancer (ES-SCLC).
The study included patients with histologically or cytologically confirmed diagnosis of ES-SCLC who had not received any prior systemic treatment for ES-SCLC and ECOG performance status 0 or 1.
A total of 457 patients were randomised (1:1) to receive:
- Arm tislelizumab + chemotherapy: tislelizumab 200 mg plus carboplatin AUC 5 mg/mL/min or cisplatin 75 mg/m² on Day 1 and etoposide 100 mg/m² intravenously on Days 1, 2, and 3 of each 21-day cycle for a maximum of 4 cycles.
- Arm placebo + chemotherapy: placebo plus carboplatin AUC 5 mg/mL/min or cisplatin 75 mg/m² on Day 1 and etoposide 100 mg/m² intravenously on Days 1, 2, and 3 of each 21-day cycle for a maximum of 4 cycles.
The choice of platinum agent (cisplatin or carboplatin) was at the investigator's discretion. Tislelizumab 200 mg monotherapy or placebo continued every 3 weeks until disease progression, loss of clinical benefit, unacceptable toxicity.
Randomisation was stratified by ECOG performance status (0 versus 1), investigator-chosen chemotherapy (carboplatin versus cisplatin), and brain metastasis (yes versus no).
The primary efficacy endpoint was overall survival (OS) in the intent-to-treat analysis set. The secondary efficacy endpoints included investigator-assessed progression-free survival (PFS), objective response rate (ORR), and duration of response (DoR) per RECIST v1.1.
Demographics and baseline characteristics were generally balanced between the 2 treatment arms. The baseline characteristics for all 457 randomised patients were: median age of 62 years (range: 31 to 78 years); 37.2% were ≥65 years of age; 81.4% male; 100% Asian (all enrolled in China), 84.9% with ECOG PS of 1; 1.1% had a history of brain metastases; 79% received carboplatin per investigator's choice; 62.6% were current smokers; and 89.3% had disease Stage IV defined by AJCC 7th Edition.
At the time of the prespecified final analysis (data cut-off 19 April 2023), BGB-A317-312 showed a statistically significant improvement in OS for patients randomised to the tislelizumab plus chemotherapy arm as compared to the placebo plus chemotherapy arm. The stratified HR was 0.75 (95% CI: 0.61, 0.93; 1-sided p-value of 0.004), with a median OS of 15.5 months in the tislelizumab plus chemotherapy arm compared to 13.5 months in the placebo plus chemotherapy arm.
A descriptive updated analysis (data cut-off 29 December 2023) showed consistent efficacy results with the final analysis. The median OS follow-up times by reverse Kaplan-Meier methodology were 39.8 months (95% CI: 36.2 to 41.4 months) in the tislelizumab plus chemotherapy arm and 36.4 months (95% CI: 35.0 to 40.9 months) in the placebo plus chemotherapy arm.
Efficacy results of the updated analysis are shown in Table 8 and Figure 8. Data for patients with brain metastases are too limited to draw conclusions on this population.
Table 8. Efficacy results in BGB-A317-312 – Updated analysis:
| Tislelizumab + Chemotherapy (N=227) | Placebo + Chemotherapy (N=230) | |
|---|---|---|
| Overall Survival | ||
| Deaths, n (%) | 175 (77.1) | 195 (84.8) |
| Median (months) (95% CI)a | 15.5 (13.5, 17.1) | 13.5 (12.1, 14.9) |
| Stratified Hazard Ratio (95% CI)b | 0.78 (0.63, 0.95) | |
| Progression-Free Survival | ||
| Events, n (%) | 178 (78.4) | 207 (90.0) |
| Median (months) (95% CI)a | 4.7 (4.3, 5.5) | 4.3 (4.2, 4.4) |
| Stratified Hazard Ratio (95% CI)b | 0.65 (0.53, 0.80) | |
| Overall Response Ratec, (%) (95% CI)d | 68.3 (61.8, 74.3) | 61.7 (55.1, 68.0) |
| Median Duration of Response (Months)c (95% CI)a | 4.3 (4.1, 5.6) | 3.7 (3.0, 4.1) |
a Median was estimated using Kaplan-Meier method with 95% CIs estimated using the method of Brookmeyer and Crowley with log-log transformation.
b Hazard ratio and 95% CI were estimated using a Cox regression model stratified by ECOG performance (1 vs 0) and platinum (Carboplatin vs Cisplatin) with placebo + chemotherapy as the reference group.
c Objective responses were confirmed per RECIST v1.1.
d The 95% CI was estimated using the Clopper-Pearson method.
Figure 8. Kaplan-Meier plot of OS in BGB-A317-312:
Gastric or gastroesophageal junction (G/GEJ) adenocarcinoma
First-line treatment of G/GEJ adenocarcinoma: BGB-A317-305
BGB-A317-305 is a randomised, multicentre, double-blind, placebo-controlled phase III study comparing the efficacy and safety of tislelizumab plus platinum and fluoropyrimidine-based chemotherapy versus placebo plus platinum and fluoropyrimidine-based chemotherapy as first-line treatment in patients with locally advanced unresectable or metastatic G/GEJ adenocarcinoma.
The study included only patients with histologically confirmed adenocarcinoma and with no prior systemic therapy for advanced disease. Patients may have received prior neoadjuvant or adjuvant therapy as long as it was completed and have no recurrence or disease progression for at least 6 months.
Patients were enrolled regardless of their tumour PD-L1 expression level, which was evaluated prospectively at a central laboratory by Tumour Area Positivity (TAP) score, which is defined as total percentage of tumor area (tumor and any desmoplastic stroma) covered by tumor cells with PD-L1 membrane staining (any intensity), and tumor associated immune cells with PD-L1 staining (any intensity), visually estimated by pathologists using Ventana PD-L1 (SP263) assay.
The study excluded patients who had squamous cell or undifferentiated or other histological type G/GEJ cancer and patients who had known HER-2 positive tumours.
Randomisation was stratified by geographical region (China [including Taiwan] versus Japan and South Korea versus rest of the world [ROW, including US and Europe]), PD-L1 expression (PD-L1 TAP score ≥5% versus PD-L1 TAP score <5%), presence of peritoneal metastasis (yes versus no) and ICC option (oxaliplatin plus capecitabine versus cisplatin plus 5-FU).
Patients were randomised (1:1) to receive tislelizumab 200 mg or placebo every 3 weeks in combination with platinum and fluoropyrimidine-based chemotherapy on a 21-day cycle. Tislelizumab (or placebo) was administered until disease progression or unacceptable toxicity. After 24 months of treatment, study therapy could be continued beyond two years if the investigator considered this to be in the best interest of the patient based on an assessment of clinical benefit and potential risks.
Chemotherapy consisted of:
- oxaliplatin 130 mg/m² IV on day 1 and capecitabine 1 000 mg/m² orally twice daily for 14 consecutive days, repeated every 3 weeks. Oxaliplatin was administered for up to 6 cycles and capecitabine was administered as maintenance therapy at investigator's discretion until disease progression or unacceptable toxicity.
or
- cisplatin 80 mg/m² IV on day 1, and 5-FU 800 mg/m²/day by continuous IV infusion over 24 hours daily on days 1 to 5, repeated every 3 weeks. Cisplatin and 5-FU were given for up to 6 cycles.
The primary efficacy endpoints were overall survival (OS) in the PD-L1 Positive Analysis Set (PD-L1 TAP score ≥5%) and ITT analysis set (all randomized patients). The secondary efficacy endpoints were PFS, ORR and DoR, as assessed by the investigator per RECIST v1.1, and health-related quality of life (HRQoL).
Tumour assessment was performed approximately every 6 weeks during the first 48 weeks and thereafter approximately every 9 weeks.
A total of 997 patients were randomised to either the tislelizumab + chemotherapy arm (n=501) or the placebo + chemotherapy arm (n=496). Of the 997 patients, 546 (54.8%) had PD-L1 TAP score ≥5% (tislelizumab + chemotherapy: n=274; placebo + chemotherapy: n=272), 931 (93.4%) received oxaliplatin + capecitabine treatment (tislelizumab + chemotherapy: n=466; placebo + chemotherapy: n=465).
In patients whose tumours expressed PD-L1 with a TAP score ≥5%, the baseline characteristics for the study population were: median age of 62 years (range: 23 to 84), 39.2% age 65 years or older; 72.2% male; 23.1% White and 73.8% Asian; 33.7% with ECOG PS of 0 and 66.3% with ECOG PS of 1. A total of 79.9% patients had primary tumour location of stomach; 98.5% of patients had metastatic disease at baseline; 43.6% and 39.7% and patients had liver metastasis and peritoneal metastasis, respectively.
At prespecified interim analysis, BGB-A317-305 demonstrated a statistically significant improvement in OS for patients randomised to the tislelizumab + chemotherapy arm as compared to the placebo + chemotherapy arm in patients with PD-L1 TAP score ≥5%. The stratified HR was 0.74 (95% CI: 0.59 to 0.94; 1-sided p-value of 0.0056), with a median OS of 17.2 months in the tislelizumab + chemotherapy arm compared to 12.6 months in the placebo + chemotherapy arm. The study also demonstrated a statistically significant improvement in PFS in patients with PD-L1 TAP score ≥5%. The stratified HR was 0.67 (95% CI: 0.55 to 0.83; 1-sided p-value <0.0001), with a median PFS of 7.2 months for tislelizumab + chemotherapy compared to 5.9 months for placebo + chemotherapy.
At prespecified final analysis, BGB-A317-305 demonstrated a statistically significant improvement for all randomized patients. The stratified HR was 0.80 (95% CI: 0.70 to 0.92; 1-sided p-value of 0.0011), with a median OS of 15.0 months in the tislelizumab + chemotherapy arm compared to 12.9 months in the placebo + chemotherapy arm. The updated results of OS in patients with PD-L1 TAP score ≥5% were consistent with its primary analysis results.
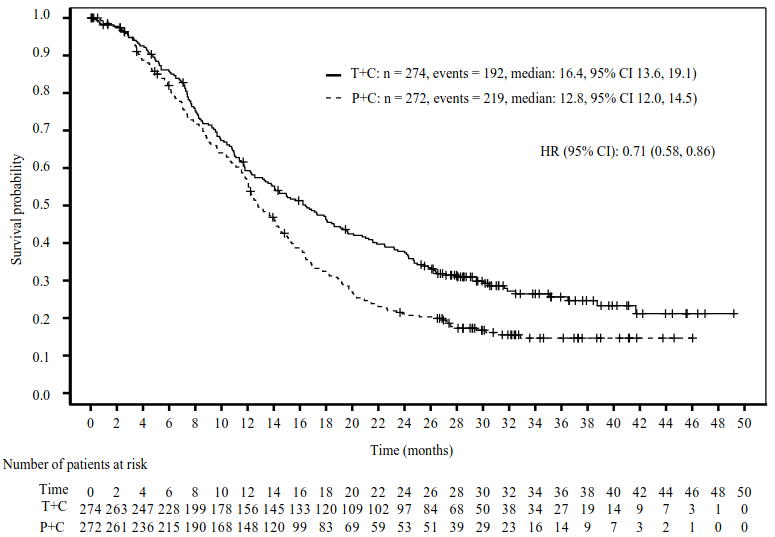
The final analysis efficacy results from patients with PD-L1 TAP score ≥5% are shown in Table 9 and in Figure 9.
Table 9. Efficacy results in BGB-A317-305 patients with PD-L1 TAP score ≥5% (final analysis):
| Tislelizumab + chemotherapy (N=274) | Placebo + chemotherapy (N=272) | |
|---|---|---|
| Patients with PD-L1 score ≥5% | ||
| Median study follow-up (months)a | 32.5 | 32.2 |
| OS | ||
| Death, n (%) | 192 (70.1) | 219 (80.5) |
| Medianb (months) (95% CI) | 16.4 (13.6, 19.1) | 12.8 (12.0, 14.5) |
| Hazard ratioc (95% CI) | 0.71 (0.58, 0.86) | |
| p-valuec,d | 0.0003e | |
| PFS | ||
| Disease progression or death, n (%) | 189 (69.0) | 216 (79.4) |
| Medianb (months) (95% CI) | 7.2 (5.8, 8.4) | 5.9 (5.6, 7.0) |
| Hazard ratioc (95% CI) | 0.68 (0.56, 0.83) | |
| ORR (%) (95% CI) | 51.5 (45.4, 57.5) | 42.6 (36.7, 48.8) |
OS = overall survival; CI = confidence interval; PFS = progression-free survival; ORR = objective response rate.
a Median follow-up time was estimated by the reverse Kaplan-Meier method.
b Medians were estimated using Kaplan-Meier method with 95% CIs estimated using the method of Brookmeyer and Crowley.
c Stratified by regions (east Asia versus US, Europe) and peritoneal metastasis.
d One-sided p-value from stratified log-rank test.
e Nominal p-value.
Figure 9. Kaplan-Meier plot of OS in BGB-A317-305 patients with PD-L1 TAP score ≥5% (final analysis):
T+C = Tislelizumab + Chemotherapy, P+C = Placebo + Chemotherapy
Both log-rank and Cox regression model were stratified by regions (east Asia versus US, Europe) and presence of peritoneal metastasis.
Oesophageal squamous cell carcinoma (OSCC)
First-line treatment of OSCC: BGB-A317-306
BGB-A317-306 is a randomised, double-blind placebo-controlled, global phase III study to compare the efficacy of tislelizumab in combination with platinum-based chemotherapy versus placebo in combination with platinum-based chemotherapy in patients with unresectable, locally advanced recurrent or metastatic OSCC.
The study enrolled patients who were not amenable to chemoradiation or surgery with curative intent. Patients were enrolled regardless of their tumour PD-L1 expression level. Where available, the archival/fresh tumour tissue specimens taken were retrospectively tested for PD-L1 expression status. PD-L1 expression was evaluated using TAP (tumour area positivity) score, defined as the total percentage of the tumour area (tumour and any desmoplastic stroma) covered by tumour cells with PD-L1 membrane staining at any intensity and tumour-associated immune cells with PD-L1 staining at any intensity, as visually estimated using the VENTANA PD-L1 (SP263) Assay.
Patients who had received prior systemic therapy for advanced or metastatic disease were excluded. A treatment-free interval of at least 6 months was required if the patient had received prior neoadjuvant/adjuvant therapy with platinum-based chemotherapy.
The study excluded patients who had evidence of fistula or complete oesophageal obstruction not amenable to treatment.
Randomisation was stratified by geographical region (Asia [excluding Japan] versus Japan versus rest of world [ROW]), prior definitive therapy (yes versus no) and investigator choice of chemotherapy (ICC; platinum with fluoropyrimidine or platinum with paclitaxel).
Patients were randomised (1:1) to receive either tislelizumab 200 mg or placebo every 3 weeks in combination with investigator's choice of chemotherapy (ICC) on a 21-day cycle. The chemotherapy doublet regimen consisted of:
- platinum (cisplatin [60 to 80 mg/m² IV on day 1] or oxaliplatin [130 mg/m² IV on day 1]) and a fluoropyrimidine (5-FU [750 to 800 mg/m² IV on days 1 to 5] or capecitabine [1000 mg/m² orally twice daily on days 1 to 14]), or
- platinum (cisplatin [60 to 80 mg/m² IV on day 1 or 2] or oxaliplatin [130 mg/m² IV on day 1 or 2]) and paclitaxel (175 mg/m² IV on day 1).
Patients were treated with tislelizumab in combination with chemotherapy or placebo in combination with chemotherapy until disease progression, as assessed by the investigator per RECIST version 1.1 or unacceptable toxicity. After 24 months of treatment, study therapy could be continued beyond two years if the investigator considered this to be in the best interest of the patient based on an assessment of clinical benefit and potential risks.
The tumour assessments were conducted every 6 weeks for the first 48 weeks, and every 9 weeks thereafter.
The primary efficacy endpoint was overall survival (OS) in the intent-to-treat (ITT) population. Secondary efficacy endpoints were progression-free survival (PFS), objective response rate (ORR) and duration of response (DoR) as assessed by the investigator per RECIST v1.1, OS in the PD-L1 positive (PD-L1 TAP score ≥10%) subgroup and health-related quality of life (HRQoL).
A total of 649 patients were randomised to receive tislelizumab in combination with chemotherapy (n=326) or placebo in combination with chemotherapy (n=323). Of the 649 patients, 293 (45.1%) patients received platinum + fluoropyrimidine, 358 patients had PD-L1 TAP score ≥5%, 184 patients had PD-L1 TAP score <5% and 107 patients had PD-L1 status unknown.
In patients whose tumours expressed PD-L1 with a TAP score ≥ 5%, the baseline characteristics were: median age 63.0 years (range: 40 to 84), 44.7% age 65 years or older; 84.9% male; 20.9% White and 78.2% Asian. 87.7% had metastatic disease at study entry and 12.3% had locally advanced disease. All patients had histological confirmation of squamous cell carcinoma. Baseline ECOG performance status was 0 (29.9%) or 1 (70.1%).
As of the data cut-off date of interim analysis (28 February 2022), BGB-A317-306 showed a statistically significant improvement in OS for all randomised patients. The stratified HR was 0.66 (95% CI, 0.54-0.80, 1-sided p-value of <0.0001), with a median OS of 17.2 months for the tislelizumab with chemotherapy arm vs. 10.6 months for the placebo with chemotherapy arm.
An updated analysis (up to 3-year follow-up; data cut-off date of 24 November 2023) showed consistent efficacy results with the interim analysis. The median follow-up times by reverse Kaplan-Meier methodology were 44.2 months in the tislelizumab in combination with chemotherapy arm and 43.8 months in the placebo in combination with chemotherapy arm.
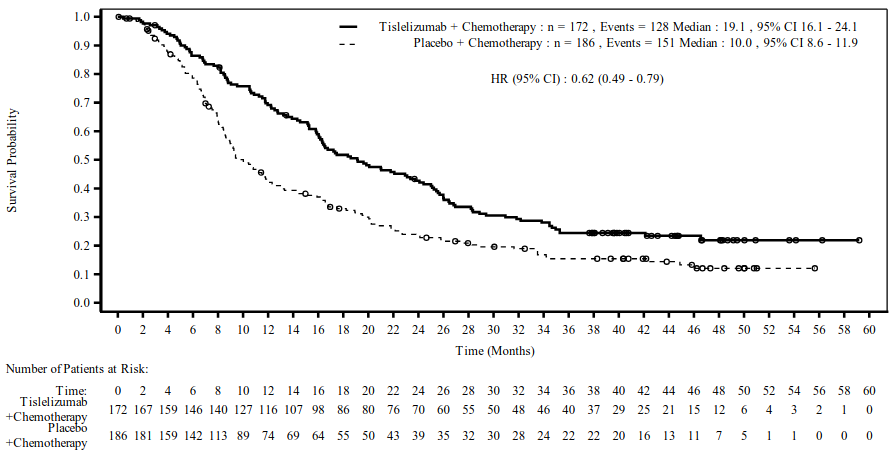
Efficacy results for patients with PD-L1 TAP score ≥5%, at 3-year follow-up, are shown in Table 10 and Figure 10.
Table 10. Efficacy results in BGB-A317-306 patients with PD-L1 TAP score ≥5% - 3-year follow-up (data cut-off 24 November 2023):
| Endpoint | Tislelizumab + chemotherapy (N=172) | Placebo + chemotherapy (N=186) |
|---|---|---|
| OS | ||
| Deaths, n (%) | 128 (74.4) | 151 (81.2) |
| Median (months) (95% CI) | 19.1 (16.1, 24.1) | 10.0 (8.6, 11.9) |
| HR (95% CI)a | 0.62 (0.49, 0.79) | |
| p-valueb | <0.0001 | |
| PFS | ||
| Events, n (%) | 119 (69.2) | 153 (82.3) |
| Median (months) (95% CI) | 8.2 (7.0, 9.8) | 5.5 (4.3, 6.4) |
| HR (95% CI)a | 0.50 (0.39, 0.65) | |
| p-valueb | <0.0001 | |
| ORR % (95% CI)c | 64.0 (56.3, 71.1) | 36.0 (29.1, 43.4) |
OS = overall survival; CI = confidence interval; HR = hazard ratio; PFS = progression-free survival; ORR = objective response rate
a Based on a stratified Cox regression model.
b One-sided nominal p-value from a stratified log rank test.
c Exact Clopper-Person-2-sided confidence interval.
Figure 10. Kaplan-Meier plot of OS in BGB-A317-306 patients with PD-L1 TAP score ≥5% - 3-year follow-up (data cut-off 24 November 2023):
Hazard ratio was based on a stratified Cox regression model.
Previously treated OSCC: BGB-A317-302
BGB-A317-302 was a randomised, controlled, open-label, global phase III study to compare the efficacy of tislelizumab versus chemotherapy in patients with unresectable, recurrent, locally advanced or metastatic OSCC who progressed on or after prior systemic treatment. Patients were enrolled regardless of their tumour PD-L1 expression level. Where available, the archival/fresh tumour tissue specimens taken were retrospectively tested for PD-L1 expression status. PD-L1 expression was evaluated at a central laboratory using the Ventana PD-L1 (SP263) assay that identified PD-L1 staining on both tumour and tumour-associated immune cells.
The study excluded patients with prior anti-PD-1/PD-L1 inhibitor treatment and tumour invasion into organs located adjacent to the oesophageal disease site (e.g. aorta or respiratory tract).
Randomisation was stratified by geographical region (Asia [excluding Japan] versus Japan versus USA/EU), ECOG PS (0 versus 1) and investigator choice of chemotherapy (ICC) option (paclitaxel versus docetaxel versus irinotecan). The choice of ICC was determined by the investigator before randomisation.
Patients were randomised (1:1) to receive tislelizumab 200 mg every 3 weeks or investigator's choice of chemotherapy (ICC), selected from the following, all given intravenously:
- paclitaxel 135 to 175 mg/m² on day 1, given every 3 weeks (also at doses of 80 to 100 mg/m² on a weekly schedule according to local and/or country-specific guidelines for standard of care), or
- docetaxel 75 mg/m² on day 1, given every 3 weeks, or
- irinotecan 125 mg/m² on days 1 and 8, given every 3 weeks.
Patients were treated with Tevimbra or one of the ICC until disease progression as assessed by the investigator per RECIST version 1.1 or unacceptable toxicity.
The tumour assessments were conducted every 6 weeks for the first 6 months, and every 9 weeks thereafter.
The primary efficacy endpoint was overall survival (OS) in the intent-to-treat (ITT) population. Secondary efficacy endpoints were OS in the PD-L1 Positive Analysis Set (PD-L1 score of visually-estimated Combined Positive Score, now known as Tumour Area Positivity [TAP] PD-L1 score 31 ≥10%), objective response rate (ORR), progression-free survival (PFS) and duration of response (DoR), as assessed by the investigator per RECIST v1.1.
A total of 512 patients were enrolled and randomised to tislelizumab (N=256) or ICC (N=256; paclitaxel [N=85], docetaxel [N=53] or irinotecan [N=118]). Of the 512 patients, 142 (27.7%) had PD-L1 score ≥10%, 222 (43.4%) had PD-L1 score <10%, and 148 (28.9%) had unknown baseline PD-L1 status.
The baseline characteristics for the study population were median age 63 years (range: 35 to 86), 39.5% age 65 years or older; 84% male; 19% White and 80% Asian; 25% with ECOG PS of 0 and 75% with ECOG PS of 1. Ninety-five percent of the study population had metastatic disease at study entry. All patients had received at least one prior anti-cancer chemotherapy, which was a platinum-based combination chemotherapy for 97% of patients.
At the time of the prespecified final analysis, BGB-A317-302 showed a statistically significant improvement in OS for patients randomised to the tislelizumab arm as compared to the ICC arm. The stratified HR was 0.70 (95% CI: 0.57, 0.85; 1-sided p-value of 0.0001), with a median OS of 8.6 months (95% CI: 7.5, 10.4) in the tislelizumab arm compared to 6.3 months (95% CI: 5.3, 7.0) in the ICC arm. The median follow-up times by reverse Kaplan-Meier methodology were 20.8 months in the tislelizumab arm and 21.1 months in the ICC arm.
An updated analysis with additional 24 months follow-up after the prespecified final analysis showed consistent efficacy results with the final analysis. The median follow-up times by reverse Kaplan-Meier methodology were 44.7 months in the tislelizumab arm and 44.0 months in the ICC arm.
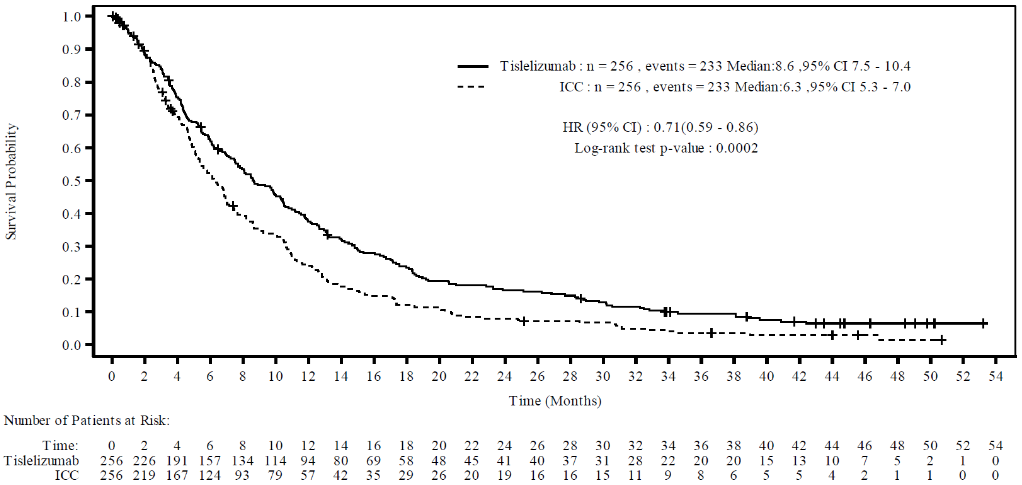
Efficacy results of the updated analysis are shown in Table 11 and Figure 11.
Table 11. Efficacy results in BGB-A317-302 – Updated analysis:
| Endpoint | Tevimbra (N=256) | Chemotherapy (N 256) |
|---|---|---|
| OS | ||
| Deaths, n (%) | 233 (91.0) | 233 (91.0) |
| Median (months)a (95% CI) | 8.6 (7.5, 10.4) | 6.3 (5.3, 7.0) |
| Hazard ratio (95% CI)b | 0.71 (0.59, 0.86) | |
| p-valuec | p=0.0002 | |
| PFS assessed by investigatord | ||
| Disease progression or death, n (%) | 229 (89.5) | 181 (70.7) |
| Median (months) (95% CI) | 1.6 (1.4, 2.7) | 2.1 (1.5, 2.7) |
| Hazard ratio (95% CI) | 0.82 (0.67, 1.01) | |
| ORR with confirmation by investigatord | ||
| ORR (%) (95% CI) | 15.2 (11.1, 20.2) | 6.6 (3.9, 10.4) |
| Median duration of response with confirmation by investigator (months) (95% CI) | 11.3 (6.5, 14.4) | 6.3 (2.8, 8.5) |
OS = overall survival; CI = confidence interval; PFS = progression-free survival; ORR = objective response rate
a Estimated using Kaplan-Meier method.
b Based on Cox regression model including treatment as covariate, and stratified by baseline ECOG status and investigator's choice of chemotherapy.
c Nominal one-sided p-value based on a log-rank test stratified by ECOG performance status and investigator's choice of chemotherapy.
d Based on ad hoc analysis.
Figure 11. Kaplan-Meier plot of OS in BGB-A317-302 (ITT analysis set) – updated analysis:

Nominal one-sided p-value is based on a log-rank test stratified by ECOG performance status and investigator's choice of chemotherapy.
Efficacy and PD-L1 subgroups (Updated analysis):
At the updated analysis of OS in the PD-L1 positive subgroup (PD-L1 score ≥10%), the stratified HR for OS was 0.54 (95% CI: 0.36 to 0.79. The median survival was 10.2 months (95% CI: 8.5 to 14.5 months) and 5.1 months (95% CI: 3.8 to 8.2 months) for the tislelizumab and ICC arms, respectively.
In the PD-L1 negative subgroup (PD-L1 score <10%), the stratified HR for OS was 0.86 (95% CI: 0.65 to 1.14), with median overall survival of 7.5 months (95% CI: 5.5 to 8.9 months) and 5.8 months (95% CI: 4.8 to 6.9 months) for the tislelizumab and ICC arms, respectively.
Nasopharyngeal carcinoma (NPC)
First-line treatment of recurrent or metastatic NPC: BGB-A317-309
BGB-A317-309 was a randomised, multicentre, double-blind, placebo-controlled phase III study to compare the efficacy and safety of tislelizumab in combination with gemcitabine and cisplatin versus placebo in combination with gemcitabine and cisplatin as first-line treatment in patients with recurrent or metastatic NPC.
Patients were treatment-naive for recurrent or metastatic NPC. A treatment-free interval of at least 6 months was required if the patient had received prior neoadjuvant chemotherapy, adjuvant chemotherapy, radiotherapy, or chemoradiotherapy with curative intent for nonmetastatic disease. The study excluded patients with local recurrence suitable for curative surgery or radiotherapy, and patients who received prior therapies targeting PD-1 or PD-L1.
Patients were randomised (1:1) to receive either tislelizumab 200 mg every 3 weeks or placebo in combination with cisplatin 80 mg/m² on Day 1 plus gemcitabine 1 g/m² on Day 1 and Day 8 of each 21-day cycle for 4 to 6 cycles. Randomised patients were stratified by gender and liver metastasis status.
Tislelizumab or placebo was administered until disease progression or unacceptable toxicity. Patients in the placebo arm were given the option to crossover to receive tislelizumab monotherapy after IRC-confirmed disease progression.
The primary efficacy endpoint was progression-free survival (PFS) as assessed by the IRC per RECIST v1.1 in the intent-to-treat (ITT) analysis set. The secondary efficacy endpoints included overall survival (OS), PFS as assessed by the investigator, objective response rate (ORR) and duration of response (DoR) as assessed by the IRC.
A total of 263 patients were randomised to receive either tislelizumab in combination with gemcitabine and cisplatin (N=131) or placebo in combination with gemcitabine and cisplatin (N=132).
The baseline characteristics for the study population were: median age of 50 years (range: 23 to 74 years), 91.6% of patients were younger than 65 years old; 78.3% of patients were male; 63.1% had ECOG PS score of 1; 100% were Asian (from China, Thailand, and Taiwan); and 46.7% were current or former smokers. 95.1% of the study population had metastatic disease at randomisation, with histological subtypes of NPC including 86.3% non-keratinised, 6.5% keratinised squamous carcinoma, and 7.2% unclassified NPC. The majority (76%) of patients had Epstein-Barr virus (EBV) DNA level ≥500 IU/mL. The baseline characteristics were generally balanced between the 2 arms.
At the time of the prespecified interim analysis (data cut-off date of 26-Mar-2021), BGB-A317-309 demonstrated a statistically significant improvement in PFS for patients randomised to tislelizumab in combination with gemcitabine and cisplatin arm compared with the placebo plus gemcitabine and cisplatin arm. The stratified HR was 0.52 (95% CI: 0.38, 0.73; 1 sided p-value of <0.0001), with a median PFS of 9.2 months in the tislelizumab plus chemotherapy arm compared to 7.4 months in the placebo plus chemotherapy arm.
An updated analysis (data cut-off date of 08-Dec-2023) showed consistent efficacy results with the interim analysis (Table 12 and Figure 12). At this time, 52.3% of patients in the control arm had crossed over to receive tislelizumab monotherapy. The median OS follow-up times by reverse Kaplan-Meier method were 41.4 months in the tislelizumab plus chemotherapy arm and 40.8 months in the placebo plus chemotherapy arm.
Data from NPC patients aged 65 years or older are too limited to draw conclusions in this population.
Table 12. Efficacy results in BGB-A317-309 (ITT Analysis Set) – Updated Analysis:
| Endpoint | Tislelizumab + Chemotherapy (N=131) | Placebo + Chemotherapy (N=132) |
|---|---|---|
| PFS by IRC | ||
| Events, n (%) | 95 (72.5) | 106 (80.3) |
| Median PFS (months) (95% CI)a | 9.6 (7.6, 11.6) | 7.4 (5.6, 7.6) |
| Stratified Hazard Ratio (95% CI)b | 0.53 (0.39, 0.71) | |
| OS | ||
| Deaths, n (%) | 55 (42.0) | 64 (48.5) |
| Median (months) (95% CI)a | 45.3 (33.4, NE) | 31.8 (25.0, NE) |
| Stratified Hazard Ratio (95% CI)b | 0.73 (0.51, 1.05) | |
Abbreviations: NE = not estimable; OS = overall survival; CI = confidence interval; PFS = progression-free survival.
a Medians were estimated by Kaplan-Meier method with 95% CIs estimated using the method of Brookmeyer and Crowley.
b Stratified by gender (male versus female) and liver metastases status (with versus without).
Figure 12. Kaplan-Meier plot of PFS in BGB-A317-309 by IRC (ITT Analysis Set) – Updated Analysis:

* Chemotherapy = Gemcitabine + Cisplatin
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with tislelizumab in all subsets of the paediatric population in the treatment of malignant neoplasms (except central nervous system, haematopoietic and lymphoid tissue) (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetics (PK) of tislelizumab were assessed for Tevimbra both as monotherapy and in combination with chemotherapy.
The PK of tislelizumab were characterised using population PK analysis with concentration data from 2596 patients with advanced malignancies who received tislelizumab doses of 0.5 to 10 mg/kg every 2 weeks, 2.0 and 5.0 mg/kg body weight every 3 weeks, and 200 mg every 3 weeks.
The time to reach 90% steady-state level is approximately 84 days (12 weeks) after 200 mg doses once every 3 weeks, and the steady-state accumulation ratio of tislelizumab PK exposure is approximately 2-fold.
Absorption
Tislelizumab is administered intravenously and therefore is immediately and completely bioavailable.
Distribution
A population pharmacokinetic analysis indicates that the steady-state volume of distribution is 6.42 l, which is typical of monoclonal antibodies with limited distribution.
Biotransformation
Tislelizumab is expected to be degraded into small peptides and amino acids via catabolic pathways.
Elimination
Based on population PK analysis, the clearance of tislelizumab was 0.153 l/day with an inter-individual variability of 26.3% and the geometrical mean terminal half-life was approximately 23.8 days with a coefficient variation (CV) of 31%.
Linearity/non-linearity
At the dosing regimens of 0.5 mg/kg to 10 mg/kg once every 2 or 3 weeks (including 200 mg once every 3 weeks and at 400 mg once every 6 weeks), the PK of tislelizumab were observed to be linear and the exposure was dose proportional.
Special populations
The effects of various covariates on tislelizumab PK were assessed in population PK analyses. The following factors had no clinically relevant effect on the exposure of tislelizumab: age (range 18 to 90 years), weight (range 32 to 130 kg), gender, race (White, Asian and other), mild to moderate renal impairment (creatinine clearance [CLCr] ≥30 ml/min), mild to moderate hepatic impairment (total bilirubin ≤3 times ULN and any AST), and tumour burden.
Renal impairment
No dedicated studies of tislelizumab have been conducted in patients with renal impairment. In the population PK analyses of tislelizumab, no clinically relevant differences in the clearance of tislelizumab were found between patients with mild renal impairment (CLCr 60 to 89 ml/min, N=1 046) or moderate renal impairment (CLCr 30 to 59 ml/min, n=320) and patients with normal renal function (CLCr ≥90 ml/min, n=1 223). Mild and moderate renal impairment had no effect on the exposure of tislelizumab (see section 4.2). Based on the limited number of patients with severe renal impairment (n=5), the effect of severe renal impairment on the pharmacokinetics of tislelizumab is not conclusive.
Hepatic impairment
No dedicated studies of tislelizumab have been conducted in patients with hepatic impairment. In the population PK analyses of tislelizumab, no clinically relevant differences in the clearance of tislelizumab were found between patients with mild hepatic impairment (bilirubin ≤ ULN and AST >ULN or bilirubin >1.0 to 1.5 x ULN and any AST, n=396) or moderate hepatic impairment (bilirubin >1.5 to 3 x ULN and any AST; n=12), compared to patients with normal hepatic function (bilirubin ≤ ULN and AST = ULN, n=2 182) (see section 4.2). Based on the limited number of patients with severe hepatic impairment (bilirubin >3 x ULN and any AST, n=2), the effect of severe hepatic impairment on the pharmacokinetics of tislelizumab is unknown.
5.3. Preclinical safety data
In repeat-dose toxicology studies in cynomolgus monkeys with intravenous dose administration at doses of 3, 10, 30 or 60 mg/kg every 2 weeks for 13 weeks (7 dose administrations), no apparent treatment-related toxicity or histopathological changes were observed at doses up to 30 mg/kg every 2 weeks, corresponding to 4.3 to 6.6 times the exposure in humans with the clinical dose of 200 mg.
No developmental and reproductive toxicity studies or animal fertility studies have been conducted with tislelizumab.
No studies have been performed to assess the potential of tislelizumab for carcinogenicity or genotoxicity.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.