ULTOMIRIS Concentrate for solution for infusion Ref.[50087] Active ingredients: Ravulizumab
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Alexion Europe SAS, 103-105 rue Anatole France, 92300 Levallois-Perret, FRANCE
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, selective immunosuppressants
ATC code: L04AA43
Mechanism of action
Ravulizumab is a monoclonal antibody IgG2/4K that specifically binds to the complement protein C5, thereby inhibiting its cleavage to C5a (the proinflammatory anaphylatoxin) and C5b (the initiating subunit of the membrane attack complex [MAC or C5b-9]) and preventing the generation of the C5b-9. Ravulizumab preserves the early components of complement activation that are essential for opsonisation of microorganisms and clearance of immune complexes.
Pharmacodynamic effects
Following ravulizumab treatment in both adult and paediatric complement inhibitor-naïve patients and eculizumab-experienced patients with PNH in Phase 3 studies, immediate, complete and sustained inhibition of serum free C5 (concentration of <0.5 μg/mL) was observed by the end of the first infusion and sustained throughout the entire 26-week treatment period in all patients. Immediate and complete inhibition of serum free C5 was also observed in adult and paediatric patients with aHUS, in adult patients with gMG, and in adult patients with NMOSD by the end of the first infusion and throughout the primary treatment period.
The extent and duration of the pharmacodynamic response in patients with PNH, aHUS, gMG, or NMOSD were exposure dependent for ravulizumab. Free C5 levels less than 0.5 μg/mL were correlated with maximal intravascular haemolysis control and complete terminal complement inhibition. In gMG, terminal complement activation leads to MAC deposition at the neuromuscular junction and impairment of neuromuscular transmission. In NMOSD, terminal complement activation leads to MAC formation and C5a-dependent inflammation, astrocyte necrosis, and damage to surrounding glial cells and neurons.
Clinical efficacy and safety
Paroxysmal nocturnal haemoglobinuria (PNH) The safety and efficacy of ravulizumab in adult patients with PNH were assessed in two open-label, randomised, active-controlled Phase 3 trials:
- a complement inhibitor-naïve study in adult patients with PNH who were naïve to complement inhibitor treatment,
- an eculizumab-experienced study in adult patients with PNH who were clinically stable after having been treated with eculizumab for at least the previous 6 months.
Ravulizumab was dosed in accordance with the recommended dosing described in section 4.2 (4 infusions of ravulizumab over 26 weeks) while eculizumab was administered according to the approved dosing regimen of eculizumab of 600 mg every week for the first 4 weeks and 900 mg every 2 weeks (15 infusions over 26 weeks).
Patients were vaccinated against meningococcal infection prior to or at the time of initiating treatment with ravulizumab or eculizumab or received prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination.
There were no noteworthy differences in the demographic or baseline characteristics between the ravulizumab and eculizumab treatment groups in either of the Phase 3 studies. The 12-month transfusion history was similar between ravulizumab and eculizumab treatment groups within each of the Phase 3 studies.
Study in complement inhibitor-naïve adult patients with PNH (ALXN1210-PNH-301)
The complement inhibitor-naïve study was a 26-week, multicentre, open-label, randomised, active-controlled, Phase 3 study conducted in 246 patients who were naïve to complement inhibitor treatment prior to study entry and was followed by a long-term extension period where all patients received ravulizumab. Eligible patients to enter this trial had to demonstrate high disease activity, defined as LDH level ≥1.5 × upper limit of normal (ULN) at screening along with the presence of 1 or more of the following PNH-related signs or symptoms within 3 months of screening: fatigue, haemoglobinuria, abdominal pain, shortness of breath (dyspnoea), anaemia (haemoglobin <10 g/dL), history of a major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction; or history of packed red blood cell (pRBC) transfusion due to PNH.
More than 80% of patients in both treatment groups had a history of transfusion within 12 months of study entry. The majority of the complement inhibitor-naïve study population was highly haemolytic 14 at baseline; 86.2% of enrolled patients presented with elevated LDH ≥3 × ULN, which is a direct measurement of intravascular haemolysis, in the setting of PNH.
Table 10 presents the baseline characteristics of the PNH patients enrolled in the complement inhibitor-naïve study, with no apparent clinically meaningful differences observed between the treatment arms.
Table 10. Baseline characteristics in the complement inhibitor-naïve study:
| Parameter | Statistics | Ravulizumab (N=125) | Eculizumab (N=121) |
|---|---|---|---|
| Age (years) at PNH diagnosis | Mean (SD) Median Min, max | 37.9 (14.90) 34.0 15, 81 | 39.6 (16.65) 36.5 13, 82 |
| Age (years) at first infusion in study | Mean (SD) Median Min, max | 44.8 (15.16) 43.0 18, 83 | 46.2 (16.24) 45.0 18, 86 |
| Sex (n, %) | Male Female | 65 (52.0) 60 (48.0) | 69 (57.0) 52 (43.0) |
| Pre-treatment LDH levels | Mean (SD) Median | 1633.5 (778.75) 1513.5 | 1578.3 (727.06) 1445.0 |
| Number of patients with packed red blood cell (pRBC) transfusions within 12 months prior to first dose | n (%) | 103 (82.4) | 100 (82.6) |
| Units of pRBC transfused within 12 months prior to first dose | Total Mean (SD) Median | 925 9.0 (7.74) 6.0 | 861 8.6 (7.90) 6.0 |
| Total PNH RBC clone size | Median | 33.6 | 34.2 |
| Total PNH granulocyte clone size | Median | 93.8 | 92.4 |
| Patients with any PNH conditionsa prior to informed consent | n (%) | 121 (96.8) | 120 (99.2) |
| Anaemia | 103 (82.4) | 105 (86.8) | |
| Haematuria or haemoglobinuria | 81 (64.8) | 75 (62.0) | |
| Aplastic anaemia | 41 (32.8) | 38 (31.4) | |
| Renal failure | 19 (15.2) | 11 (9.1) | |
| Myelodysplastic syndrome | 7 (5.6) | 6 (5.0) | |
| Pregnancy complication | 3 (2.4) | 4 (3.3) | |
| Otherb | 27 (21.6) | 13 (10.7) |
a Based on medical history.
b "Other" as specified on case report form included thrombocytopenia, chronic kidney disease, and pancytopenia, as well as a number of other conditions.
The coprimary endpoints were transfusion avoidance, and haemolysis as directly measured by normalisation of LDH levels (LDH levels ≤1 × ULN; the ULN for LDH is 246 U/L). Key secondary endpoints included the percent change from baseline in LDH levels, change in quality of life (FACIT-Fatigue), the proportion of patients with breakthrough haemolysis and proportion of patients with stabilised haemoglobin.
Ravulizumab was non-inferior compared to eculizumab for both coprimary endpoints, avoidance of pRBC transfusion per protocol-specified guidelines and LDH normalisation from day 29 to day 183, and for all 4 key secondary endpoints (Figure 1).
Figure 1. Analysis of coprimary and secondary endpoints – full analysis set (complement inhibitor-naïve study):
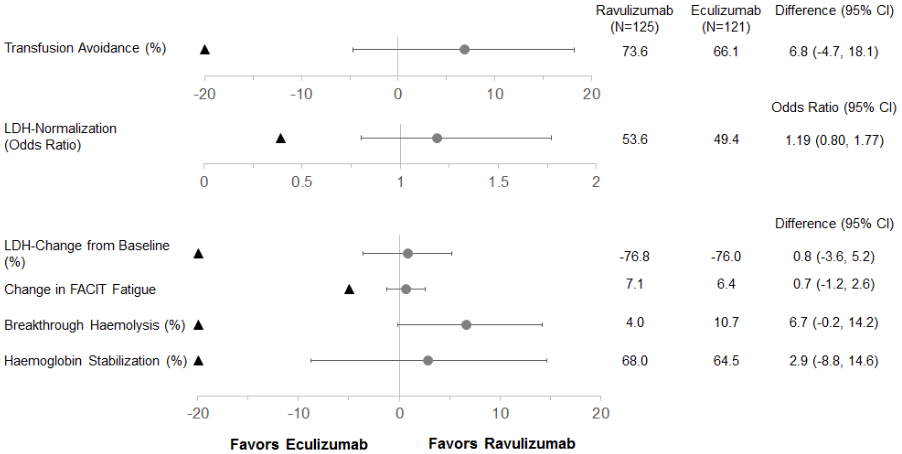
Note: The black triangle indicates the non-inferiority margins, and grey dots indicates point estimates.
Note: LDH = lactate dehydrogenase; CI = confidence interval; FACIT = Functional Assessment of Chronic Illness Therapy.
The final efficacy analysis for the study included all patients ever treated with ravulizumab (n=244) and had a median treatment duration of 1423 days. The final analysis confirmed that ravulizumab treatment responses observed during the Primary Evaluation Period were maintained throughout the duration of the study.
Study in adult PNH patients previously treated with eculizumab (ALXN1210-PNH-302)
The eculizumab-experienced study was a 26-week, multicentre, open-label, randomised, active-controlled Phase 3 study conducted in 195 patients with PNH who were clinically stable (LDH ≤1.5 x ULN) after having been treated with eculizumab for at least the past 6 months and was followed by a long-term extension period where all patients received ravulizumab.
PNH medical history was similar between ravulizumab and eculizumab treatment groups. The 12-month transfusion history was similar between ravulizumab and eculizumab treatment groups and more than 87% of patients in both treatment groups had not received a transfusion within 12 months of study entry. The mean total PNH RBC clone size was 60.05%, mean total PNH granulocyte clone size was 83.30%, and the mean total PNH monocyte clone size was 85.86%.
Table 11 presents the baseline characteristics of the PNH patients enrolled in the eculizumab-experienced study, with no apparent clinically meaningful differences observed between the treatment arms.
Table 11. Baseline characteristics in the eculizumab-experienced study:
| Parameter | Statistics | Ravulizumab (N=97) | Eculizumab (N=98) |
|---|---|---|---|
| Age (years) at PNH diagnosis | Mean (SD) Median Min, max | 34.1 (14.41) 32.0 6, 73 | 36.8 (14.14) 35.0 11, 74 |
| Age (years) at first infusion in study | Mean (SD) Median Min, max | 46.6 (14.41) 45.0 18, 79 | 48.8 (13.97) 49.0 23, 77 |
| Sex (n, %) | Male Female | 50 (51.5) 47 (48.5) | 48 (49.0) 50 (51.0) |
| Pre-treatment LDH levels | Mean (SD) Median | 228.0 (48.71) 224.0 | 235.2 (49.71) 234.0 |
| Number of patients with pRBC/whole blood transfusions within 12 months prior to first dose | n (%) | 13 (13.4) | 12 (12.2) |
| Units of pRBC/whole blood transfused within 12 months prior to first dose | Total Mean (SD) Median | 103 7.9 (8.78) 4.0 | 50 4.2 (3.83) 2.5 |
| Patients with any PNH conditionsa prior to informed consent | n (%) | 90 (92.8) | 96 (98.0) |
| Anaemia | 64 (66.0) | 67 (68.4) | |
| Haematuria or haemoglobinuria | 47 (48.5) | 48 (49.0) | |
| Aplastic anaemia | 34 (35.1) | 39 (39.8) | |
| Renal failure | 11 (11.3) | 7 (7.1) | |
| Myelodysplastic syndrome | 3 (3.1) | 6 (6.1) | |
| Pregnancy complication | 4 (4.1) | 9 (9.2) | |
| Otherb | 14 (14.4) | 14 (14.3) |
a Based on medical history.
b "Other" category included neutropenia, renal dysfunction, and thrombopenia, as well as a number of other conditions.
The primary endpoint was haemolysis as measured by LDH percent change from baseline. Secondary endpoints included the proportion of patients with breakthrough haemolysis, quality-of-life (FACIT-Fatigue), transfusion avoidance (TA), and proportion of patients with stabilised haemoglobin.
Ravulizumab was non-inferior compared to eculizumab for the primary endpoint, percent change in LDH from baseline to day 183, and for all 4 key secondary endpoints (Figure 2).
Figure 2. Analysis of primary and secondary endpoints – full analysis set (eculizumab-experienced study):
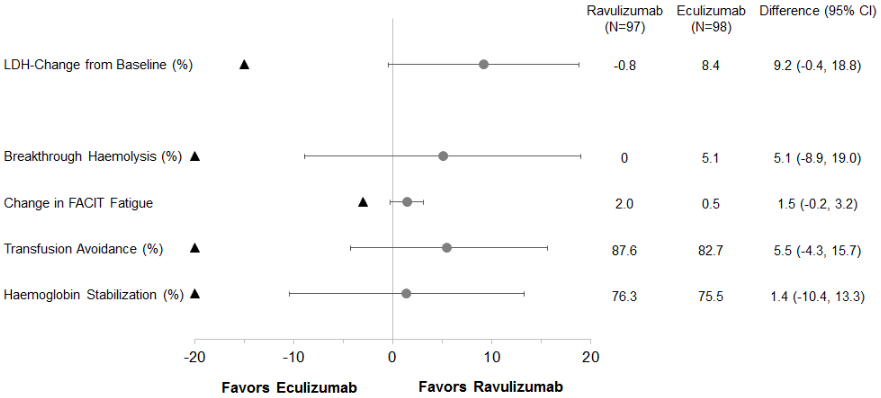
Note: The black triangle indicates the non-inferiority margins, and grey dot indicates point estimates.
Note: LDH = lactate dehydrogenase; CI = confidence interval.
The final efficacy analysis for the study included all patients ever treated with ravulizumab (n=192) and had a median treatment duration of 968 days. The final analysis confirmed that ravulizumab treatment responses observed during the primary evaluation period were maintained throughout the duration of the study.
Atypical haemolytic uremic syndrome (aHUS)
Study in adult patients with aHUS (ALXN1210-aHUS-311)
The adult study was a multicentre, single arm, Phase 3 study conducted in patients with documented aHUS who were naïve to complement inhibitor treatment prior to study entry and had evidence of thrombotic microangiopathy (TMA). The study consisted of a 26-week initial evaluation period and patients were allowed to enter an extension period for up to 4.5 years. A total of 58 patients with documented aHUS were enrolled. Enrolment criteria excluded patients presenting with TMA due to thrombotic thrombocytopenic purpura (TTP) or Shiga toxin Escherichia coli related haemolytic uremic syndrome (STEC-HUS). Two patients were excluded from the full analysis set due to a confirmed diagnosis of STEC-HUS. Ninety-three percent of patients had extra renal signs (cardiovascular, pulmonary, central nervous system, gastrointestinal, skin, skeletal muscle) or symptoms of aHUS at baseline.
Table 12 presents the demographics and baseline characteristics of the 56 adult patients enrolled in Study ALXN1210-aHUS-311 that constituted the full analysis set.
Table 12. Baseline characteristics in the adult study:
| Parameter | Statistics | Ravulizumab (N=56) |
|---|---|---|
| Age at time of first infusion (years) | Mean (SD) Min, max | 42.2 (14.98) 19.5, 76.6 |
| Sex Male | n (%) | 19 (33.9) |
| Race Asian White Other | n (%) | 15 (26.8) 29 (51.8) 12 (21.4) |
| History of transplant | n (%) | 8 (14.3) |
| Platelets (109/L) blood | n Median (min,max) | 56 95.25 (18, 473) |
| Haemoglobin (g/L) blood | n Median (min,max) | 56 85.00 (60.5, 140) |
| LDH (U/L) serum | n Median (min,max) | 56 508.00 (229.5, 3249) |
| eGFR (mL/min/1.73 m²) | n (%) Median (min,max) | 55 10.00 (4, 80) |
| Patients on dialysis | N (%) | 29 (51.8) |
| Patients post partum | N (%) | 8 (14.3) |
Note: Percentages are based on the total number of patients.
Abbreviations: eGFR = estimated glomerular filtration rate; LDH = lactate dehydrogenase; max = maximum; min = minimum.
The primary endpoint was Complete TMA Response during the 26-week Initial Evaluation Period, as evidenced by normalisation of haematological parameters (platelet count ≥150 x 109/L and LDH ≤246 U/L) and ≥25% improvement in serum creatinine from baseline. Patients had to meet each Complete TMA Response criteria at 2 separate assessments obtained at least 4 weeks (28 days) apart, and any measurement in between.
Complete TMA Response was observed in 30 of the 56 patients (53.6%) during the 26-week initial evaluation period as shown in Table 13.
Table 13. Complete TMA response and complete TMA response components analysis during the 26-week initial evaluation period (ALXN1210-aHUS-311):
| Total | Responder | ||
|---|---|---|---|
| n | Proportion (95% CI)a | ||
| Complete TMA Response | 56 | 30 | 0.536 (0.396, 0.675) |
| Components of Complete TMA Response | |||
| Platelet count normalisation | 56 | 47 | 0.839 (0.734, 0.944) |
| LDH normalisation | 56 | 43 | 0.768 (0.648, 0.887) |
| ≥25% improvement in serum creatinine from baseline | 56 | 33 | 0.589 (0.452, 0.727) |
| Haematologic normalisation | 56 | 41 | 0.732 (0.607, 0.857) |
a 95% CIs for the proportion were based on the asymptotic Gaussian approximation method with a continuity correction.
Abbreviations: CI = confidence interval; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy.
Four additional patients had a Complete TMA Response that was confirmed after the 26-week initial evaluation period (with a Complete TMA Response occurring at Days 169, 302, 401 and 407). resulting in an overall Complete TMA Response in 34 of 56 patients (60.7%; 95% CI: 47.0%, 74.4%). Individual component response increased to 48 (85.7%; 95% CI: 75.7%, 95.8%) patients for platelet count normalisation, 47 (83.9%; 95% CI: 73.4%, 94.4%) patients for LDH normalisation, and 35 (62.5%; 95% CI: 48.9%, 76.1%) patients for renal function improvement.
19 Complete TMA Response was achieved at a median time of 86 days (7 to 169 days). An increase in mean platelet count was observed rapidly after commencement of ravulizumab, increasing from 118.52 × 109/L at baseline to 240.34 × 109/L at Day 8 and remaining above 227 × 109/L at all subsequent visits in the initial evaluation period (26 weeks). Similarly, mean LDH value decreased from baseline over the first 2 months of treatment and was sustained over the duration of the initial evaluation period (26 weeks).
Of the patients who presented at CKD Stage 5, 67.6% (23/34) showed an improvement of 1 or more CKD Stages. Chronic kidney disease stage continued to improve for many patients (19/30) after achieving Complete TMA Response during the 26-week initial evaluation period. 17 of the 29 patients who required dialysis at study entry were able to discontinue dialysis by the end of the available follow-up while 6 of 27 patients who were off dialysis at baseline were on dialysis at last available follow-up. Table 14 summarises the secondary efficacy outcomes for Study ALXN1210-aHUS-311.
Table 14. Secondary efficacy outcome for study ALXN1210-aHUS-311:
| Parameters | Study ALXN1210-aHUS-311 (N=56) | |
|---|---|---|
| Haematologic TMA parameters, Day 183 Platelets (109/L) blood Mean (SD) Median LDH (U/L) serum Mean (SD) Median | Observed value (n=48) 237.96 (73.528) 232.00 194.46 (58.099) 176.50 | Change from baseline (n=48) 114.79 (105.568) 125.00 -519.83 (572.467) -310.75 |
| Increase in haemoglobin of ≥20 g/L from baseline with a confirmatory result through Initial Evaluation Period m/n proportion (95% CI)* | 40/56 0.714 (0.587, 0.842) | |
| CKD stage shift from baseline, Day 183 Improveda m/n Proportion* (95% CI) Worsenedb m/n Proportion* (95% CI) | 32/47 0.681 (0.529, 0.809) 2/13 0.154 (0.019, 0.454) | |
| eGFR (mL/min/1.73 m²), Day 183 Mean (SD) Median | Observed value (n=48) 51.83 (39.162) 40.00 | Change from baseline (n=47) 34.80 (35.454) 29.00 |
Note: n: number of patients with available data for specific assessment at Day 183 visit. m: number of patients meeting specific criterion. Chronic kidney disease (CKD) stage is classified based on the National Kidney Foundation Chronic Kidney Disease Stage. Stage 5 is considered the worst category, while Stage 1 is considered the best category. Baseline is derived based on the last available eGFR before starting treatment.
Improved/Worsened: compared to CKD stage at baseline.
* 95% confidence intervals (95% CIs) are based on
exact confidence limits using the Clopper-Pearson method.
a Excludes those with CKD Stage 1 at baseline as they cannot improve. bExcludes patients with Stage 5 at baseline as they cannot worsen.
Abbreviations: eGFR = estimated glomerular filtration rate; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy.
Generalised Myasthenia Gravis (gMG)
Study in adult patients with gMG
The efficacy and safety of ravulizumab in adult patients with gMG was assessed in a Phase 3, randomised, double-blind, placebo-controlled, multicentre study (ALXN1210-MG-306). Patients participating in this study were subsequently allowed to enter an Open-Label Extension Period during which all patients received ravulizumab.
Patients with gMG (diagnosed for at least 6 months) with a positive serologic test for anti-acetylcholine receptor (AChR) antibodies, MGFA (Myasthenia Gravis Foundation of America) clinical classification Class II to IV and remaining symptomatology as evidenced by a Myasthenia Gravis Activities of Daily Living (MG-ADL) total score ≥ 6 were randomised to receive either ravulizumab (N=86) or placebo (N=89). Patients on immunosuppressant therapies (corticosteroids, azathioprine, cyclophosphamide, cyclosporine, methotrexate, mycophenolate mofetil, or tacrolimus) were permitted to continue on therapy throughout the course of the study. In addition, rescue therapy (including high dose corticosteroid, PE/PP, or IVIg) was allowed if a patient experienced clinical deterioration, as defined by the study protocol.
A total of 162 (92.6%) patients completed the 26-week Randomised-Controlled Period of Study ALXN1210-MG-306. The baseline characteristics of patients are presented in Table 15. The majority (97%) of patients included in the study had been treated with at least one immunomodulatory therapy including immunosuppressant therapies, PE/PP, or IVIg in the last two years prior to enrolment.
Table 15. Baseline disease characteristics in study ALXN1210-MG-306:
| Parameter | Statistics | Placebo (N=89) | Ravulizumab (N=86) |
|---|---|---|---|
| Sex Male Female | n (%) | 44 (49.4) 45 (50.6) | 42 (48.8) 44 (51.2) |
| Age at first dose of study drug (years) | Mean (SD) (min, max) | 53.3 (16.05) (20, 82) | 58.0 (13.82) (19, 79) |
| Elderly (≥65 years of age) at study entry | n (%) | 24 (27.0) | 30 (34.9) |
| Duration of MG since diagnosis (years) | Mean (SD) (min, max) Median | 10.0 (8.90) (0.5, 36.1) 7.6 | 9.8 (9.68) (0.5, 39.5) 5.7 |
| Baseline MG-ADL Score | Mean (SD) (min, max) Median | 8.9 (2.30) (6.0, 15.0) 9.0 | 9.1 (2.62) (6.0, 24.0) 9.0 |
| Baseline QMG Score | Mean (SD) (min, max) Median | 14.5 (5.26) (2.0, 27.0) 14.0 | 14.8 (5.21) (6.0, 39.0) 15.0 |
| Baseline MGFA classification Class II (mild weakness) Class III (moderate weakness) Class IV (severe weakness) | n (%) | 39 (44) 45 (51) 5 (6) | 39 (45) 41 (48) 6 (7) |
| Any prior intubation since diagnosis (MGFA Class V) | n (%) | 9 (10.1) | 8 (9.3) |
| Number of patients with prior MG crisis since diagnosisa | n (%) | 17 (19.1) | 21 (24.4) |
| Number of stable immunosuppressant therapiesb at study entry 0 1 ≥2 | n (%) | 8 (9.0) 34 (38.2) 47 (52.8) | 10 (11.6) 40 (46.5) 36 (41.9) |
a Prior MG crisis information was collected as part of medical history and not evaluated as per the clinical protocol definition.
b Immunosuppressant therapies include corticosteroids, azathioprine, cyclophosphamide, cyclosporine, methotrexate, mycophenolate mofetil, or tacrolimus.
Abbreviations: Max = maximum; min = minimum; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living; MGFA = Myasthenia Gravis Foundation of America; QMG = Quantitative Myasthenia Gravis; SD = standard deviation
The primary endpoint was the change from baseline to Week 26 in the MG-ADL total score.
The secondary endpoints, also assessed changes from baseline to Week 26, included the change in the Quantitative Myasthenia Gravis (QMG) total score, the proportion of patients with improvements of at 21 least 5 and 3 points in the QMG and MG-ADL total scores, respectively, as well as changes in quality-of-life assessments.
Ravulizumab demonstrated a statistically significant change in the MG-ADL total score as compared to placebo. Primary and secondary endpoint results are presented in Table 16.
Table 16. Analysis of primary and secondary efficacy endpoints:
| Efficacy Endpoints at Week 26 | Placebo (N=89) LS Mean (SEM) | Ravulizumab (N=86) LS Mean (SEM) | Statistic for Comparison | Treatment Effect (95% CI) | p-value (Using Mixed Effect Repeated Measures) |
|---|---|---|---|---|---|
| MG-ADL | -1.4 (0.37) | -3.1 (0.38) | Difference in change from baseline | -1.6 (-2.6, -0.7) | 0.0009 |
| QMG | -0.8 (0.45) | -2.8 (0.46) | Difference in change from baseline | -2.0 (-3.2, -0.8) | 0.0009 |
| MG-QoL15r | -1.6 (0.70) | -3.3 (0.71) | Difference in change from baseline | -1.7 (-3.4, 0.1) | 0.0636 |
| Neuro-QoL-fatigue | -4.8 (1.87) | -7.0 (1.92) | Difference in change from baseline | -2.2 (-6.9, 2.6) | 0.3734a |
a The endpoint was not formally tested for statistical significance; a nominal p-value was reported.
Abbreviations: CI = confidence interval; LS = least squares; MG-ADL = Myasthenia Gravis Activities of Daily Living; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item scale; Neuro-QoL-fatigue = Neurological Quality of Life Fatigue; QMG = Quantitative Myasthenia Gravis; SEM = standard error of mean.
In Study ALXN1210-MG-306, a clinical responder in the MG-ADL total score was defined as having at least a 3-point improvement. The proportion of clinical responders at Week 26 was 56.7% on ravulizumab compared with 34.1% on placebo (nominal p=0.0049). A clinical responder in the QMG total score was defined as having at least a 5-point improvement. The proportion of clinical responders at Week 26 was 30.0% on ravulizumab compared with 11.3% on placebo (p=0.0052).
Table 17 presents an overview of the patients with clinical deterioration and patients requiring rescue therapy over the 26-week Randomised-Controlled Period.
Table 17. Clinical deterioration and rescue therapy:
| Variable | Statistic | Placebo (N=89) | Ravulizumab (N=86) |
|---|---|---|---|
| Total number of patients with clinical deterioration | n (%) | 15 (16.9) | 8 (9.3) |
| Total number of patients requiring rescue therapya | n (%) | 14 (15.7) | 8 (9.3) |
a Rescue therapy included high-dose corticosteroid, plasma exchange/plasmapheresis, or intravenous immunoglobulin.
At the time of the analysis, 150 of the 158 patients who entered the Open-Label Extension Period were ongoing in the study.
In patients who initially received ULTOMIRIS during the Randomised-Controlled Period and continued to receive ULTOMIRIS during the first 34-weeks of the Open-Label Extension Period, the treatment effect was sustained (Figure 3). In patients who initially received placebo during the 26-week Randomised-Controlled Period and initiated treatment with ULTOMIRIS during the Open-Label Extension Period, a rapid and sustained treatment response (Figure 3), was observed.
Figure 3. Change from randomised-controlled period baseline in MG-ADL total score (A) and QMG total score (B) through week 60 (mean and 95% CI):
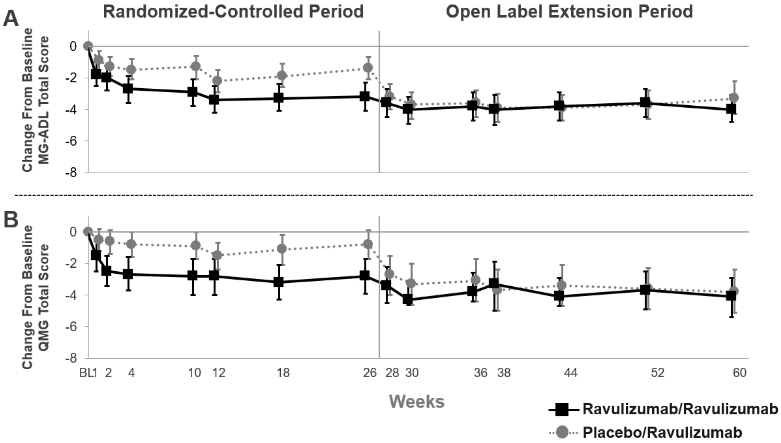
Abbreviations: CI = confidence interval; MG-ADL = Myasthenia Gravis Activities of Daily Living; QMG = Quantitative Myasthenia Gravis
In the Open-Label Extension Period of the study, clinicians had the option to adjust immunosuppressant therapies. In patients followed for 34 weeks in the Open-Label Extension Period, 28.0% of patients decreased their daily dose of corticosteroid therapy and 6.2% of patients stopped corticosteroid therapy. The most common reason for change in corticosteroid therapies was improvement in MG symptoms while on ravulizumab treatment.
Neuromyelitis Optica Spectrum Disorder (NMOSD)
Study in adult patients with NMOSD
The efficacy of ravulizumab in adult patients with anti-AQP4 antibody-positive NMOSD was assessed in a global, open-label clinical study (ALXN1210-NMO-307).
Study ALXN1210-NMO-307 enrolled 58 adult patients with NMOSD who had a positive serologic test for anti-AQP4 antibodies, at least 1 relapse in the last 12 months prior to the Screening Period, and an Expanded Disability Status Scale (EDSS) score of ≤ 7. Prior treatment with immunosuppressant therapies (ISTs) was not required for enrolment and 51.7% of patients were on ravulizumab monotherapy. Patients on selected ISTs (i.e., corticosteroids, azathioprine, mycophenolate mofetil, tacrolimus) were permitted to continue on therapy in combination with ravulizumab, with a requirement for stable dosing until they reached Week 106 in the study. In addition, acute therapy for relapse treatment (including high-dose corticosteroids, PE/PP, and IVIg) was allowed if a patient experienced a relapse during the study.
Patients included in the study had a mean age of 47.4 years (ranging from 18 to 74 years) and most of them were female (90%). Median age at NMOSD initial clinical presentation was of 42.5 years, ranging from 16 to 73 years. Baseline disease characteristics are shown in Table 18.
Table 18. Patient disease history and baseline characteristics in study ALXN1210-NMO-307:
| Variable | Statistic | ALXN1210-NMO-307 Ravulizumab (N=58) |
|---|---|---|
| Time from NMOSD initial clinical presentation to first dose of study drug (years) | Mean (SD) | 5.2 (6.38) |
| Median | 2.0 | |
| Min, max | 0.19, 24.49 | |
| Historical ARR within 24 months prior to screening | Mean (SD) | 1.87 (1.59) |
| Median | 1.44 | |
| Min, max | 0.5, 6.9 | |
| Baseline HAI score | Mean (SD) | 1.2 (1.42) |
| Median | 1.0 | |
| Min, max | 0, 7 | |
| Baseline EDSS score | Mean (SD) | 3.30 (1.58) |
| Median | 3.25 | |
| Min, max | 0.0, 7.0 | |
| Any historical rituximab use | n (%) | 21 (36.2) |
| Number of patients receiving stable corticosteroids only at study entry | n (%) | 12 (20.7) |
| Number of patients not receiving any IST at study entry | n (%) | 30 (51.7) |
Abbreviations: ARR = annualised relapse rate; EDSS = Expanded Disability Status Scale; HAI = Hauser Ambulation Index; IST = immunosuppressant therapy; Max = maximum; Min = minimum; NMOSD = neuromyelitis optica spectrum disorder; SD = standard deviation.
The primary endpoint of Study ALXN1210-NMO-307 was the time to first adjudicated on-trial relapse as determined by an independent adjudication committee. No adjudicated on-trial relapse was observed in ravulizumab-treated patients during the primary treatment period. All ravulizumab-treated patients remained relapse free over the median follow-up of 90.93 weeks. Ravulizumab-treated patients experienced consistent relapse-free primary endpoint result with or without concomitant IST treatment.
Ravulizumab has not been studied for the acute treatment of relapses in NMOSD patients.
Paediatric population
Paroxysmal nocturnal haemoglobinuria (PNH)
Study in paediatric patients with PNH (ALXN1210-PNH-304)
The paediatric study (ALXN1210-PNH-304) is a multicentre, open-label, Phase 3 study conducted in eculizumab-experienced and complement inhibitor-naïve paediatric patients with PNH. From interim results, a total of 13 PNH paediatric patients completed ravulizumab treatment during the primary evaluation period (26 weeks) of Study ALXN1210-PNH-304. Five of the 13 patients had never been treated with a complement inhibitor and 8 patients received treatment with eculizumab prior to study entry.
Most of the patients were between 12 years and 17 years of age at first infusion (mean: 14.4 years), with 2 patients under 12 years old (11 years and 9 years old). Eight of the 13 patients were female. Mean weight at baseline was 56 kg, ranging from 37 to 72 kg. Table 19 presents the baseline disease history and characteristics of the paediatric patients enrolled in Study ALXN1210-PNH-304.
Table 19. Disease history and characteristics at baseline (full analysis set):
| Variable | Complement inhibitor- naïve patients (N=5) | Eculizumab- experienced patients (N=8) |
|---|---|---|
| Total PNH RBC clone size (%) Median (min, max) | (N=4) 40.05 (6.9, 68.1) | (N=6) 71.15 (21.2, 85.4) |
| Total PNH granulocyte clone size (%) Median (Min, max) | 78.30 (36.8, 99.0) | 91.60 (20.3, 97.6) |
| Number of patients with pRBC/whole blood transfusions within 12 months prior to first dose, n (%) | 2 (40.0) | 2 (25.0) |
| Number of pRBC/whole blood transfusions within 12 months prior to first dose Total Median (min, max) | 10 5.0 (4, 6) | 2 1.0 (1, 1) |
| Units of pRBC/whole blood transfused within 12 months prior to first dose Total Median (min, max) | 14 7.0 (3, 11) | 2 2.0 (2, 2) |
| Patients with any PNH-associated conditions prior to informed consent, n (%) | 5 (100) | 8 (100) |
| Anaemia | 2 (40.0) | 5 (62.5) |
| Haematuria or haemoglobinuria | 2 (40.0) | 5 (62.5) |
| Aplastic anaemia | 3 (60.0) | 1 (12.5) |
| Renal failure | 2 (40.0) | 2 (25.0) |
| Othera | 0 | 1 (12.5) |
| Pre-treatment LDH levels (U/L) Median (min, max) | 588.50 (444, 2269.7) | 251.50 (140.5, 487) |
a Other PNH-associated conditions were reported as "renal and splenic infarcts" and "multiple lesions concerning for embolic process".
Note: Percentages were based on the total number of patients in each cohort.
Abbreviations: LDH = lactate dehydrogenase; max = maximum; min = minimum; PNH = paroxysmal nocturnal haemoglobinuria; pRBC = packed red blood cell; RBC = red blood cell.
Based on body weight, patients received a loading dose of ravulizumab on Day 1, followed by maintenance treatment on Day 15 and once every 8 weeks (q8w) thereafter for patients weighing ≥20 kg, or once every 4 weeks (q4w) for patients weighing <20 kg. For patients who entered the study on eculizumab therapy, Day 1 of study treatment was planned to occur 2 weeks from the patient's last dose of eculizumab.
The weight-based dose regimen of ravulizumab provided immediate, complete, and sustained inhibition of terminal complement throughout the 26-week primary evaluation period regardless of prior experience with eculizumab. Following initiation of ravulizumab treatment, steady-state therapeutic serum concentrations of ravulizumab were achieved immediately after the first dose and maintained throughout the 26-week primary evaluation period in both cohorts. There were no breakthrough haemolysis events in the study and no patients had post-baseline free C5 levels above 0.5 μg/mL.
Mean percent change from baseline in LDH was -47.91% on Day 183 in the complement inhibitor-naïve cohort and remained stable in the eculizumab-experienced cohort during the 26-week primary evaluation period. Sixty percent (3/5) of complement inhibitor-naïve patients and 75% (6/8) of eculizumab-experienced patients achieved haemoglobin stabilisation by Week 26 respectively. Transfusion-avoidance was reached by 84.6% (11/13) of patients during the 26-week primary evaluation period.
These interim efficacy results are presented in Table 20 below.
Table 20. Efficacy outcomes from the paediatric study in PNH patients (ALXN1210-PNH-304) - 26-week primary evaluation period:
| End Point | Ravulizumab (Naïve, N=5) | Ravulizumab (Switch, N=8) |
|---|---|---|
| LDH-Percent change from Baseline Mean (SD) | -47.91 (52.716) | 4.65 (44.702) |
| Transfusion Avoidance Percentage (95% CI) | 60.0 (14.66, 94.73) | 100.0 (63.06, 100.00) |
| Haemoglobin Stabilisation Percentage (95% CI) | 60.0 (14.66, 94.73) | 75 (34.91, 96.81) |
| Breakthrough Haemolysis (%) | 0 | 0 |
Abbreviations: LDH = lactate dehydrogenase
Long term efficacy results through end of study over a median treatment duration of 915 days resulted in a sustained treatment response in paediatric patients with PNH.
Based on data from these interim results, the efficacy of ravulizumab in paediatric PNH patients appears to be similar to that observed in adult PNH patients.
Atypical haemolytic uremic syndrome (aHUS)
Use of Ultomiris in paediatric patients for treatment of aHUS is supported by evidence from one paediatric clinical study (a total of 31 patients with documented aHUS were enrolled; 28 patients aged 10 months to 17 years were included in the full analysis set).
Study in paediatric patients with aHUS (ALXN1210 aHUS 312)
The paediatric study is a 26-week ongoing, multicentre, single arm, Phase 3 study conducted in paediatric patients.
A total of 21 eculizumab-naïve patients with documented diagnosis of aHUS and evidence of TMA were enrolled, of which 18 were included in the Full Analysis set. Enrolment criteria excluded patients presenting with TMA due to TTP and STEC-HUS. Two patients were given a single dose, and one patient received 2 doses, but then discontinued and were excluded from the full analysis set because aHUS was not confirmed. The overall mean weight at baseline was 22.2 kg; majority of the patients were in the baseline weight category ≥10 to <20 kg. The majority of patients (72.2%) had pretreatment extra renal signs (cardiovascular, pulmonary, central nervous system, gastrointestinal, skin, skeletal muscle) or symptoms of aHUS at baseline. At baseline, 33.3% (n=6) of patients had CKD Stage 5.
A total of 10 patients, who switched from eculizumab to ravulizumab, had documented diagnosis of aHUS and evidence of TMA were enrolled. Patients had to have clinical response to eculizumab prior to enrolment (i.e. LDH <1.5 X ULN and platelet count ≥150,000/μL, and eGFR >30 mL/min/1.73 m²). Consequently, there is no information on the use of ravulizumab in patient refractory to eculizumab.
Table 21 presents the baseline characteristics of the paediatric patients enrolled in Study ALXN1210-aHUS-312.
Table 21. Demographics and baseline characteristics in study ALXN1210-aHUS-312:
| Parameter | Statistics | Ravulizumab (Naïve, N=18) | Ravulizumab (Switch, N=10) |
|---|---|---|---|
| Age at time of first infusion (years) category Birth to <2 years 2 to <6 years 6 to <12 years 12 to <18 years | n (%) | 2 (11.1) 9 (50.0) 5 (27.8) 2 (11.1) | 1 (10.0) 1 (10.0) 1 (10.0) 7 (70.0) |
| Sex Male | n (%) | 8 (44.4) | 9 (90.0) |
| Racea American Indian or Alaskan Native Asian Black or African American White Unknown | n (%) | 1 (5.6) 5 (27.8) 3 (16.7) 9 (50.0) 1 (5.6) | 0 (0.0) 4 (40.0) 1 (10.0) 5 (50.0) 0 (0.0) |
| History of transplant | n (%) | 1 (5.6) | 1 (10.0) |
| Platelets (109/L) blood | Median (min, max) | 51.25 (14, 125) | 281.75 (207, 415.5) |
| Haemoglobin (g/L) | Median (min, max) | 74.25 (32, 106) | 132.0 (114.5, 148) |
| LDH (U/L) | Median (min, max) | 1963.0 (772, 4985) | 206.5 (138.5, 356) |
| eGFR (mL/min/1.73 m²) | Median (min, max) | 22.0 (10, 84) | 99.75 (54, 136.5) |
| Required dialysis at baseline | n (%) | 6 (33.3) | 0 (0.0) |
Note: Percentages are based on the total number of patients.
a Patients can have multiple races selected.
Abbreviations: eGFR = estimated glomerular filtration rate; LDH = lactate dehydrogenase; max = maximum; min = minimum.
The primary endpoint was Complete TMA Response during the 26-week Initial Evaluation Period, as evidenced by normalisation of haematological parameters (platelet ≥150 x 109/L and LDH ≤246 U/L) and ≥25% improvement in serum creatinine from baseline. Patients had to meet all Complete TMA Response criteria at 2 separate assessments obtained at least 4 weeks (28 days) apart, and any measurement in between.
Complete TMA Response was observed in 14 of the 18 naïve patients (77.8%) during the 26-week initial evaluation period as shown in Table 22.
Table 22. Complete TMA response and complete TMA response components analysis during the 26-week initial evaluation period (ALXN1210-aHUS-312):
| Total | Responder | ||
|---|---|---|---|
| n | Proportion (95% CI)a | ||
| Complete TMA Response | 18 | 14 | 0.778 (0.524, 0.936) |
| Components of Complete TMA Response | |||
| Platelet count normalisation | 18 | 17 | 0.944 (0.727, 0.999) |
| LDH normalisation | 18 | 16 | 0.889 (0.653, 0.986) |
| ≥25% improvement in serum creatinine from baseline | 18 | 15 | 0.833 (0.586, 0.964) |
| Haematologic normalisation | 18 | 16 | 0.889 (0.653, 0.986) |
Note: 1 patient withdrew from study after receiving 2 doses of ravulizumab.
a 95% CIs for the proportion were based on the asymptotic Gaussian approximation method with a continuity correction.
Abbreviations: CI = confidence interval; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy
Complete TMA Response during the initial evaluation period was achieved at a median time of 30 days (15 to 97 days). All patients with Complete TMA Response maintained it through the initial evaluation period with continuous improvements seen in renal function. An increase in mean platelet count was observed rapidly after commencement of ravulizumab, increasing from 60.50 × 109/L at baseline to 296.67 × 109/L at Day 8 and remained above 296 × 109/L at all subsequent visits in the initial evaluation period (26 weeks).
Three additional patients had a Complete TMA Response that was confirmed after the 26-week initial evaluation period (with a Complete TMA Response occurring at Days 291, 297 and 353).; thus, 17 of 18 (94.4%) paediatric patients (95% CI: 72.7%, 99.9%) had a Complete TMA Response. Individual component response increased to 17 of 18 (94.4%; 95% CI: 72.7%, 99.9%) patients for platelet count normalisation, 17 of 18 (94.4%; 95% CI: 72.7%, 99.9%) patients for LDH normalisation, and 17 of 18 (94.4%; 95% CI: 72.7%, 99.9%) patients for renal function improvement.
All 6 of the patients who required dialysis at study entry were able to discontinue dialysis; 5 of which had already done so by Day 43. No patient started dialysis during the study. The majority of the patient population (15/17), improved by 1 or more CKD stages by Day 183; 14 patients improved by 2 or more stages. Table 23 summarises the secondary efficacy results for Study ALXN1210-aHUS-312.
Table 23. Secondary efficacy outcome for study ALXN1210-aHUS-312:
| Parameters | Study ALXN1210-aHUS-312 (N=18) | |
|---|---|---|
| Haematologic TMA parameters, Day 183 Platelets (109/L) blood Mean (SD) Median LDH (U/L) serum Mean (SD) Median | Observed value (n=17) 304.94 (75.711) 318.00 262.41 (59.995) 247.00 | Change from baseline (n=17) 245.59 (91.827) 247.00 -2044.13 (1328.059) -1851.50 |
| Increase in haemoglobin of ≥20 g/L from baseline with a confirmatory result through Initial Evaluation Period m/N proportion* (95% CI) | 16/18 0.889 (0.653, 0.986) | |
| CKD stage shift from baseline, Day 183 Improveda m/n Proportion* (95% CI) Worsenedb m/n Proportion* (95% CI) | 15/17 0.882 (0.636, 0.985) 0/11 0.000 (0.000, 0.285) | |
| eGFR (mL/min/1.73 m²), Day 183 Mean (SD) Median | Observed value (n=17) 108.5 (56.87) 108.0 | Change from baseline (n=17) 85.4 (54.33) 80.0 |
Note: n: number of patients with available data for specific assessment at Day 183 visit. m: number of patients meeting specific criterion. Chronic kidney disease (CKD) stage is classified based on the National Kidney Foundation Chronic Kidney Disease Stage. Stage 1 is considered the best category, while Stage 5 is considered the worst category. Baseline is derived based on the last available eGFR before starting treatment. Improved/Worsened: Compared to CKD stage at baseline.
* 95% confidence intervals (95% CIs) are based on exact confidence limits using the Clopper Pearson method.
a Improved excludes patients with Stage 1 at baseline, as they cannot improve;
b worsened excludes patients with
Stage 5 at baseline as they cannot worsen.
Abbreviations: eGFR = estimated glomerular filtration rate; LDH = lactate dehydrogenase; TMA = thrombotic microangiopathy.
In eculizumab-experienced patients, switching to ravulizumab maintained disease control as evidenced by stable hematologic and renal parameters, with no apparent impact on safety.
The efficacy of ravulizumab for the treatment of aHUS appears similar in paediatric and adult patients.
Generalised myasthenia gravis (gMG)
The European Medicines Agency has deferred the obligation to submit the results of studies with Ultomiris in one or more subsets of the paediatric population in the treatment of myasthenia gravis. See 4.2 for information on paediatric use.
Neuromyelitis optica spectrum disorder (NMOSD)
The European Medicines Agency has deferred the obligation to submit the results of studies with Ultomiris in one or more subsets of the paediatric population in the treatment of NMOSD. See 4.2 for information on paediatric use.
5.2. Pharmacokinetic properties
Absorption
Because the route of administration is an intravenous infusion and the pharmaceutical form is a solution, 100% of the administered dose of ravulizumab is considered bioavailable. The time to maximum observed concentration (tmax) is expected at the end of infusion (EOI) or soon after EOI. Therapeutic steady-state drug concentrations are reached after the first dose.
Distribution
The mean (standard deviation [SD]) central volume and volume of distribution at steady state for adult and paediatric patients with PNH or aHUS and adult patients with gMG or NMOSD are presented in Table 24.
Biotransformation and elimination
As an immunoglobulin gamma (IgG) monoclonal antibody, ravulizumab is expected to be metabolised in the same manner as any endogenous IgG (degraded into small peptides and amino acids via catabolic pathways) and is subject to similar elimination. Ravulizumab contains only natural occurring amino acids and has no known active metabolites. The mean (SD) values for terminal elimination half-life and clearance of ravulizumab in adult and paediatric patients with PNH, adult and paediatric patients with aHUS, and adult patients with gMG or NMOSD are presented in Table 24.
Table 24. Estimated central volume, distribution, biotransformation and elimination parameters following ravulizumab administration:
| Adult and paediatric patients with PNH | Adult and paediatric patients with aHUS | Adult patients with gMG | Adult patients with NMOSD | |
|---|---|---|---|---|
| Estimated central volume (litres) Mean (SD) | Adults: 3.44 (0.66) Paediatrics: 2.87 (0.60) | Adults: 3.25 (0.61) Paediatrics: 1.14 (0.51) | 3.42 (0.756) | 2.91 (0.571) |
| Volume of distribution at steady state (litres) Mean (SD) | 5.30 (0.9) | 5.22 (1.85) | 5.74 (1.16) | 4.77 (0.819) |
| Terminal elimination half- life (days) Mean (SD) | 49.6 (9.1) | 51.8 (16.2) | 56.6 (8.36) | 64.3 (11.0) |
| Clearance (litres/day) Mean (SD) | 0.08 (0.022) | 0.08 (0.04) | 0.08 (0.02) | 0.05 (0.016) |
Abbreviations: aHUS = atypical haemolytic uremic syndrome; gMG = generalised myasthenia gravis; NMOSD = neuromyelitis optica spectrum disorder; PNH = paroxysmal nocturnal haemoglobinuria; SD = standard deviation.
Linearity/non-linearity
Over the studied dose and regimen range, ravulizumab exhibited dose proportional and time linear pharmacokinetics (PK).
Special populations
Weight
Body weight is a significant covariate in patients with PNH, aHUS, gMG, or NMOSD resulting in lower exposures in heavier patients. Weight-based dosing is proposed in section 4.2, Table 1, Table 3 and Table 4.
No formal trial of the effect of sex, race, age (geriatric), hepatic or renal impairment on the pharmacokinetics of ravulizumab was conducted. However, based on population-PK assessment no impact of sex, age, race and hepatic or renal function on ravulizumab PK was identified in the studied healthy volunteers, subjects and patients with PNH, aHUS, gMG, or NMOSD, and as a result, no dosing adjustment is considered necessary.
The pharmacokinetics of ravulizumab have been studied in aHUS patients with a range of renal impairment including patients receiving dialysis. There have been no observed differences in pharmacokinetic parameters noted in these subpopulations of patients including patients with proteinuria.
5.3. Preclinical safety data
Animal reproductive toxicology studies have not been conducted with ravulizumab but were conducted in mice with a murine surrogate complement inhibitory antibody, BB5.1. No clear treatment-related effects or adverse effects were observed in the murine surrogate reproductive toxicology studies in mice. When maternal exposure to the antibody occurred during organogenesis, two cases of retinal dysplasia and one case of umbilical hernia were observed among 230 offspring born to mothers exposed to the higher antibody dose (approximately 4 times the maximum recommended human ravulizumab dose, based on a body weight comparison); however, the exposure did not increase foetal loss or neonatal death.
No animal studies have been conducted to evaluate the genotoxic and carcinogenic potential of ravulizumab.
Non-clinical data reveal no special hazard for humans based on nonclinical studies using a murine surrogate molecule, BB5.1, in mice.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.