VAFSEO Film-coated tablet Ref.[50970] Active ingredients: Vadadustat
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: AKEBIA EUROPE Limited, 70 Sir John Rogerson's Quay, Dublin 2, Co. Dublin, D02 R296, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Anti-anaemic preparations, other anti-anaemic preparations
ATC code: B03XA08
Mechanism of action
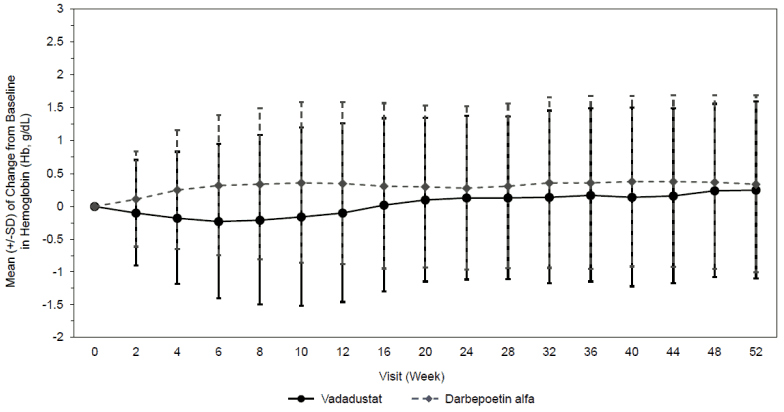
Vadadustat is a hypoxia-inducible factor prolyl-hydroxylase inhibitor which leads to increased cellular levels of hypoxia-inducible factor thereby stimulating endogenous erythropoietin (EPO) production, increasing iron mobilization and red blood cell production, resulting in gradual rate of rise in Hb (see Figures 1 and 2).
Cardiac electrophysiology
Vadadustat did not cause any clinically significant QTc prolongation following a 600 mg and 1200 mg dose in healthy subjects.
Clinical efficacy and safety
The efficacy and safety of vadadustat given once daily for the treatment of anaemia in adult patients with CKD was studied compared to darbepoetin alfa in two global multi-centre, randomised, activecontrolled, non-inferiority, open-label studies in DD patients.
The population in DD-CKD for Vafseo was 19 to 93 years of age, 55.9% male, and the percentage of Caucasian, Hispanic, Black (including African Americans) and Asian patients was 64.5%, 38.5%, 24.1%, and 4.5%, respectively.
In both studies, non-inferiority of vadadustat to darbepoetin alfa was to be concluded if the lower bound of the 95% CI for the difference in estimated mean change in average Hb from Baseline in the 2 treatment groups was greater than the prespecified non-inferiority margin of -0.75 g/dL. Patients were randomised 1:1 to receive Vafseo with a starting dose of 300 mg once daily or darbepoetin alfa administered subcutaneously or intravenously as per prescribing information for 52 weeks to assess the efficacy endpoints. Vafseo was titrated in increments/reductions of 150 mg up to 600 mg to achieve the patient's Hb target. After 52 weeks, patients were continued study treatment to assess long-term safety until the event-driven major adverse cardiovascular event (MACE) endpoints were reached. The primary efficacy endpoint for each study was the difference in mean change of Hb from baseline to the primary evaluation period (Weeks 24 to 36). The key secondary efficacy endpoint was the difference in mean change of Hb from baseline to the secondary evaluation period (Weeks 40 to 52). The primary safety endpoint was time to first MACE. MACE was defined as all-cause mortality, non-fatal myocardial infarction (MI) and non-fatal stroke.
Treatment of anaemia
The two studies INNO2VATE 1 and INNO2VATE 2 were conducted in adult DD-CKD patients with baseline Hb values between 8.0 to 11.0 g/dL in the United States (US) and 9.0 to 12.0 g/dL outside the US. INNO2VATE 1 included patients with incident DD-CKD who initiated dialysis within 16 weeks of beginning their trial participation and who were erythropoiesis-stimulating agent (ESA)-naïve, had limited prior ESA use or were maintained on ESAs. INNO2VATE 2 included patients on chronic maintenance dialysis for more than 12 weeks who had converted from prior ESA therapy. In both studies, Vafseo met the primary Hb endpoint according to predefined noninferiority margin (-0.75 g/dL). Results for the primary and secondary efficacy endpoints are provided in Table 4. Course of Hb during treatment in individual studies is provided in Figure 1 and Figure 2.
Table 4. INNO2VATE studies:
| INNO2VATE 1 | INNO2VATE 2 | |||
|---|---|---|---|---|
| Hb (g/dL) | Vafseo N=181 | Darbepoetin Alfa N=188 | Vafseo N=1777 | Darbepoetin Alfa N=1777 |
| Baseline mean (SD) | 9.37 (1.07) | 9.19 (1.14) | 10.25 (0.85) | 10.23 (0.83) |
| Primary endpoint Weeks 24 to 36 mean (SD) | 10.36 (1.13) | 10.61 (0.94) | 10.36 (1.01) | 10.53 (0.96) |
| Adjusted mean change from baseline (LSM) [95% CI] | 1.26 [1.05, 1.48] | 1.58 [1.37, 1.79] | 0.19 [0.12, 0.25] | 0.36 [0.29, 0.42] |
| Key secondary endpoint Weeks 40 to 52 mean (SD) | 10.51 (1.19) | 10.55 (1.14) | 10.40 (1.04) | 10.58 (0.98) |
| Adjusted mean change from baseline (LSM) [95% CI] | 1.42 [1.17, 1.68] | 1.50 [1.23, 1.76] | 0.23 [0.16, 0.29] | 0.41 [0.34, 0.48] |
CI: confidence interval; LSM: least squares mean; SD: standard deviation
Figure 1. Mean (+/-SD) of change from baseline in Hb (g/dL) for INNO2VATE 1 correction:
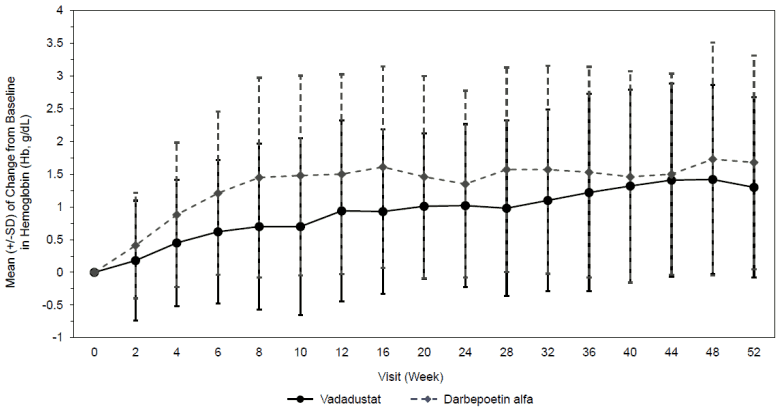
Figure 2. Mean (+/-SD) of change from baseline in Hb (g/dL) for INNO2VATE 2 conversion:
Cardiovascular outcomes
The incidence of major adverse cardiovascular events (MACE) was evaluated as part of the long-term safety evaluation of the two global efficacy studies in DD-CKD patients. Vafseo met the composite primary safety endpoint defined as non-inferiority of Vafseo to darbepoetin alfa in time to occurrence of MACE for the global study population (1.3 NI margin [HR (95% CI) was 0.96 (0.83, 1.11)]) (see Table 5).
Table 5. INNO2VATE analysis* of the composite 3-point MACE and individual cardiovascular endpoints:
| Vafseo N=1947 n (%) | Darbepoetin Alfa N=1955 n (%) | Hazard Ratio [95% CI] | |
|---|---|---|---|
| Any major adverse cardiovascular events (MACE) | 355 (18.2) | 377 (19.3) | 0.96 [0.83, 1.11] |
| All-cause mortality | 253 (13.0) | 253 (12.9) | |
| Non-fatal myocardial infarction | 76 (3.9) | 87 (4.5) | |
| Non-fatal stroke | 26 (1.3) | 37 (1.9) |
* The MACE analyses were conducted on randomised subjects who received at least 1 dose of study treatment.
CI: confidence interval; MACE: major adverse cardiovascular events.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Vafseo in one or more subsets of the paediatric population for the treatment of anaemia associated with chronic disorders (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Vadadustat is rapidly absorbed after single and repeated oral doses. Median time to peak plasma concentrations (Tmax) is approximately 2 to 3 hours.
No significant accumulation has been observed after repeated dosing in healthy subjects.
Vafseo may be administered with or without food. Administration of a 450 mg Vafseo tablet with a standard high-fat meal decreased Cmax by 27% and decreased the AUC by 6%, relative to fasted conditions.
Distribution
Vadadustat is highly protein bound (greater than or equal to 99.5% in human plasma). The mean blood to plasma ratio was less than 1 (0.50 to 0.55) suggesting minimal sequestration into red blood cells (RBCs). In patients with CKD the apparent volume of distribution (Vd/F) was 11.6 L.
Biotransformation
Vadadustat is primarily metabolised via direct glucuronidation by UDP-glucuronosyltransferase (UGT) enzymes to O-glucuronide conjugates. The major metabolite vadadustat-O-glucuronide (15% of the AUC of plasma radioactivity). Vadadustat acyl glucuronide (0.047% of the total radioactivity in plasma) is a minor metabolite. Vadadustat metabolites are not active.
Elimination
The half-life of vadadustat in DD-CKD patients was 9.2 hours. After a single oral dose of radiolabelled vadadustat 650 mg to healthy adults, 85.9% of the dose was recovered (58.9% in urine and 26.9% in faeces). The excretion for vadadustat (unchanged form) was less than 1% in urine and about 9% in faeces.
Pharmacokinetics in special populations
Renal impairment
Vadadustat exposures in DD-CKD patients were approximately 2-fold higher compared to healthy subjects. No significant differences in pharmacokinetics (Cmax, AUC or mean half-life) were observed when Vafseo was administered 4 hours before dialysis or 2 hours after dialysis.
Hepatic impairment
Moderate hepatic impairment (Child-Pugh Class B) did not significantly affect the AUC or Cmax of vadadustat compared to healthy subjects. The half-life and apparent total body clearance for vadadustat were comparable between subjects with normal hepatic function and subjects with moderate hepatic function. Vadadustat has not been studied in severe hepatic impairment (Child-Pugh Class C).
Age, gender, race, and body weight
Population pharmacokinetic analysis did not suggest any clinically significant effects of age (19 to 104 years), gender, race, or body weight (47 to 118 kg) on the pharmacokinetics of vadadustat.
A sensitivity analysis at body weight extremes (30.1 to 204 kg) showed that the dose titration algorithm resulted in predicted Hb levels at the limits of the predefined window of 10 to 12 g/dL. Therefore, no dose-adjustment is proposed at body weight extremes.
5.3. Preclinical safety data
In non-clinical trials, mortalities were observed in mice, rats, rabbits and dogs due to exaggerated pharmacological effects such as polycythemia and hyperviscosity of the blood, leading to thrombosis and organ infarct at dose levels that were clinically relevant (starting from exposure multiples of 0.04 to the maximum recommended therapeutic dose of 600 mg).
Non-clinical data reveal no other special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity or carcinogenic potential.
Vadadustat was not teratogenic in either the rat or the rabbit up to the highest dose level tested (160 mg/kg/day and 50 mg/kg/day, respectively), corresponding to 1.7 and 0.16 times the human exposure at the 600 mg dose (based on AUC in NDD-CKD patients), respectively, in the dams. Development effects were noted only in the rat at dose levels corresponding to 1.7 times the human exposure at the 600 mg dose; characterised as a decrease in foetal body weight and an increased incidence of a reduction in skeletal ossification, both of which were considered secondary to the decline in body weight and food consumption in the pregnant dams. However, in a rat dose finding study, at doses that caused significant maternal toxicity, there was an increase in postimplantation loss at ≥120 mg/kg/day and decreased foetal body weight at 240 mg/kg/day, but no teratogenicity.
Vadadustat was excreted in the milk in rats with a ratio of milk to plasma of up to 14.49.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.