VEMLIDY Film-coated tablet Ref.[7482] Active ingredients: Tenofovir alafenamide
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Gilead Sciences Ireland UC, Carrigtohill, County Cork, T45 DP77, Ireland
Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Special warnings and precautions for use
Hepatitis B Virus (HBV) transmission
Patients must be advised that this medicinal product does not prevent the risk of transmission of HBV to others through sexual contact or contamination with blood. Appropriate precautions must continue to be used.
Patients with decompensated liver disease
There are limited data on the safety and efficacy of tenofovir alafenamide in HBV infected patients with decompensated liver disease and who have a Child Pugh Turcotte (CPT) score >9 (i.e. class C). These patients may be at higher risk of experiencing serious hepatic or renal adverse reactions. Therefore, hepatobiliary and renal parameters should be closely monitored in this patient population (see section 5.2).
Exacerbation of hepatitis
Flares on treatment
Spontaneous exacerbations in chronic hepatitis B are relatively common and are characterised by transient increases in serum alanine aminotransferase (ALT). After initiating antiviral therapy, serum ALT may increase in some patients. In patients with compensated liver disease, these increases in serum ALT are generally not accompanied by an increase in serum bilirubin concentrations or hepatic decompensation. Patients with cirrhosis may be at a higher risk for hepatic decompensation following hepatitis exacerbation, and therefore should be monitored closely during therapy.
Flares after treatment discontinuation
Acute exacerbation of hepatitis has been reported in patients who have discontinued treatment for CHB, usually in association with rising HBV DNA levels in plasma. The majority of cases are self-limited but severe exacerbations, including fatal outcomes, may occur after discontinuation of treatment for CHB. Hepatic function should be monitored at repeated intervals with both clinical and laboratory follow-up for at least 6 months after discontinuation of treatment for CHB. If appropriate, resumption of CHB therapy may be warranted.
In patients with advanced liver disease or cirrhosis, treatment discontinuation is not recommended since post-treatment exacerbation of hepatitis may lead to hepatic decompensation. Liver flares are especially serious, and sometimes fatal in patients with decompensated liver disease.
Renal impairment
Patients with creatinine clearance <30 mL/min
The use of tenofovir alafenamide once daily in patients with CrCl ≥15 mL/min and <30 mL/min is based on Week 96 data on the efficacy and safety of switching from another antiviral regimen to tenofovir alafenamide in an open-label clinical study of virologically suppressed HBV-infected patients (see sections 4.8 and 5.1). There are very limited data on the safety and efficacy of tenofovir alafenamide in HBV-infected patients with CrCl <15 mL/min on chronic haemodialysis (see sections 4.8, 5.1 and 5.2).
The use of this medicinal product is not recommended in patients with CrCl < 15 mL/min who are not receiving haemodialysis (see section 4.2).
Nephrotoxicity
Post-marketing cases of renal impairment, including acute renal failure and proximal renal tubulopathy have been reported with tenofovir alafenamide-containing products. A potential risk of nephrotoxicity resulting from chronic exposure to low levels of tenofovir due to dosing with tenofovir alafenamide cannot be excluded (see section 5.3).
It is recommended that renal function is assessed in all patients prior to, or when initiating, therapy with this treatment and that it is also monitored during therapy in all patients as clinically appropriate. In patients who develop clinically significant decreases in renal function, or evidence of proximal renal tubulopathy, discontinuation of this medicinal product should be considered.
Patients co-infected with HBV and hepatitis C or D virus
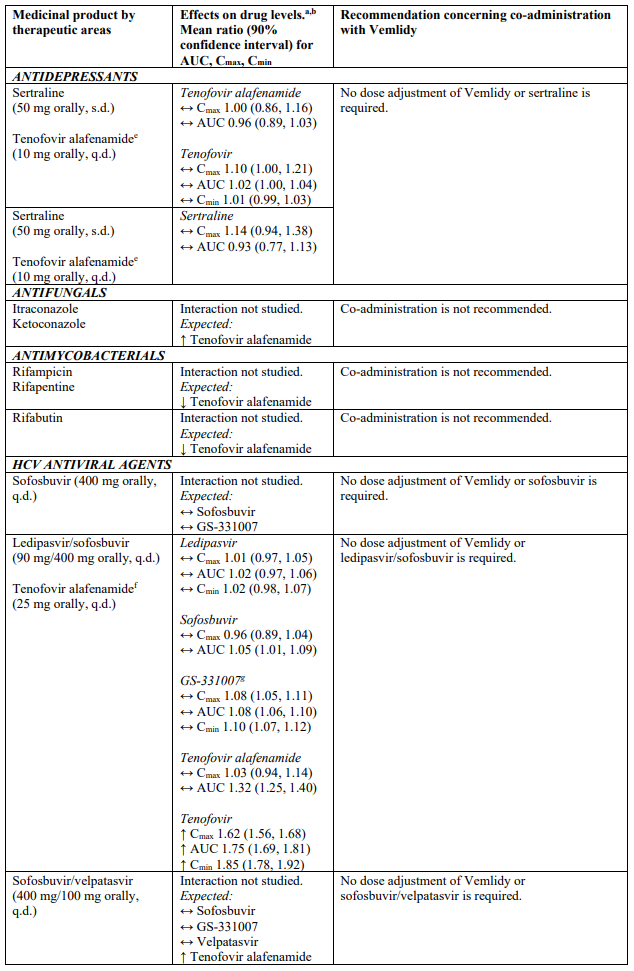
There are no data on the safety and efficacy of tenofovir alafenamide in patients co-infected with hepatitis C (HCV) or D (HDV) virus. Co-administration guidance for the treatment of HCV should be followed (see section 4.5).
HBV and Human Immunodeficiency Virus (HIV) co-infection
HIV antibody testing should be offered to all HBV infected patients whose HIV-1 infection status is unknown before initiating therapy with this medicinal product. In patients who are co-infected with HBV and HIV, Vemlidy should be co-administered with other antiretroviral medicinal products to ensure that the patient receives an appropriate regimen for treatment of HIV (see section 4.5).
Co-administration with other medicinal products
This medicinal product should not be co-administered with medicinal products containing tenofovir alafenamide, tenofovir disoproxil or adefovir dipivoxil.
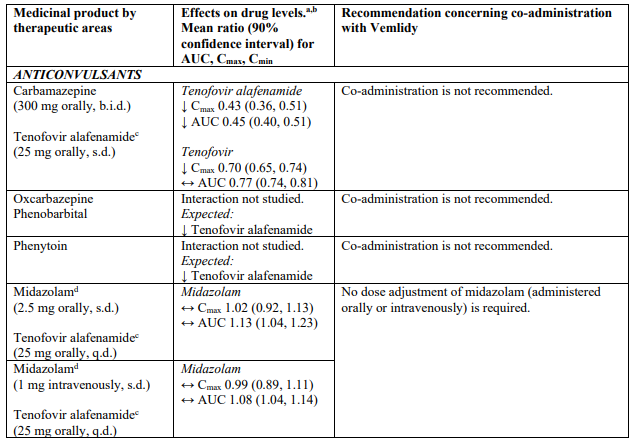
Co-administration of this treatment with certain anticonvulsants (e.g. carbamazepine, oxcarbazepine, phenobarbital and phenytoin), antimycobacterials (e.g. rifampicin, rifabutin and rifapentine) or St. John’s wort, all of which are inducers of P-glycoprotein (P-gp) and may decrease tenofovir alafenamide plasma concentrations, is not recommended.
Co-administration of this treatment with strong inhibitors of P-gp (e.g. itraconazole and ketoconazole) may increase tenofovir alafenamide plasma concentrations. Co-administration is not recommended.
Paediatric population
Reductions in bone mineral density (BMD ≥4%) of the lumbar spine and of whole body have been reported in some paediatric patients 6 years of age and older weighing at least 25 kg who received tenofovir alafenamide for 48 weeks (see sections 4.8 and 5.1). The long-term effects of changes in BMD on the growing bone, including the risk of fracture, are uncertain. A multidisciplinary approach is recommended to decide the appropriate monitoring during treatment.
Excipients with known effect
This medicinal product contains lactose monohydrate. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take this medicinal product.
This medicinal product contains less than 1 mmol sodium (23 mg) per tablet, that is to say essentially ‘sodium-free’.
Interaction with other medicinal products and other forms of interaction
Interaction studies have only been performed in adults.
This medicinal product should not be co-administered with medicinal products containing tenofovir disoproxil, tenofovir alafenamide or adefovir dipivoxil.
Medicinal products that may affect tenofovir alafenamide
Tenofovir alafenamide is transported by P-gp and breast cancer resistance protein (BCRP). Medicinal products that are P-gp inducers (e.g., rifampicin, rifabutin, carbamazepine, phenobarbital or St. John’s wort) are expected to decrease plasma concentrations of tenofovir alafenamide, which may lead to loss of therapeutic effect of Vemlidy. Co-administration of such medicinal products with tenofovir alafenamide is not recommended.
Co-administration of tenofovir alafenamide with medicinal products that inhibit P-gp and BCRP may increase plasma concentrations of tenofovir alafenamide. Co-administration of strong inhibitors of P-gp with tenofovir alafenamide is not recommended.
Tenofovir alafenamide is a substrate of OATP1B1 and OATP1B3 in vitro. The distribution of tenofovir alafenamide in the body may be affected by the activity of OATP1B1 and/or OATP1B3.
Effect of tenofovir alafenamide on other medicinal products
Tenofovir alafenamide is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 in vitro. It is not an inhibitor or inducer of CYP3A in vivo.
Tenofovir alafenamide is not an inhibitor of human uridine diphosphate glucuronosyltransferase (UGT) 1A1 in vitro. It is not known whether tenofovir alafenamide is an inhibitor of other UGT enzymes.
Drug interaction information for Vemlidy with potential concomitant medicinal products is summarised in Table 1 below (increase is indicated as “↑”, decrease as “↓”, no change as “↔”; twice daily as “b.i.d.”, single dose as “s.d.”, once daily as “q.d.”; and intravenously as “IV”). The drug interactions described are based on studies conducted with tenofovir alafenamide, or are potential drug interactions that may occur with Vemlidy.
Table 1. Interactions between Vemlidy and other medicinal products:



Fertility, pregnancy and lactation
Pregnancy
A moderate amount of data on pregnant women exposed to tenofovir alafenamide (between 300-1000 pregnancy outcomes) indicate no malformative or feto/neonatal toxicity.
Animal studies do not indicate direct or indirect harmful effects with respect to reproductive toxicity (see section 5.3).
The use of tenofovir alafenamide may be considered during pregnancy, if necessary.
Breast-feeding
Based on published data, tenofovir alafenamide and tenofovir are excreted in human milk at low levels in women administered with tenofovir alafenamide. There is insufficient information on the effects of tenofovir in newborns/infants.
A risk to the breast-fed newborns/infants cannot be excluded; therefore, tenofovir alafenamide should not be used during breast-feeding.
Fertility
No human data on the effect of tenofovir alafenamide on fertility are available. Animal studies do not indicate harmful effects of tenofovir alafenamide on fertility.
Effects on ability to drive and use machines
Vemlidy may have minor influence on the ability to drive and use machines. Patients should be informed that dizziness has been reported during treatment with tenofovir alafenamide.
Undesirable effects
Summary of the safety profile
Assessment of adverse reactions is based on clinical study data and postmarketing data. In pooled safety data from 2 controlled Phase 3 studies (GS-US-320-0108 and GS-US-320-0110; “Study 108” and “Study 110”, respectively), the most frequently reported adverse reactions at Week 96 analysis were headache (12%), nausea (6%), and fatigue (6%). After Week 96, patients either remained on their original blinded treatment up to Week 144 or received open-label tenofovir alafenamide.
The safety profile of tenofovir alafenamide was similar in virologically suppressed patients switching from tenofovir disoproxil to tenofovir alafenamide in Study 108, Study 110 and a controlled Phase 3 study GS-US-320-4018 (Study 4018). Changes in lipid laboratory tests were observed in these studies following a switch from tenofovir disoproxil (see section 5.1).
Tabulated summary of adverse reactions
The following adverse drug reactions have been identified with tenofovir alafenamide in patients with CHB (Table 2). The adverse reactions are listed below by body system organ class and frequency based on the Week 96 analysis. Frequencies are defined as follows: very common (≥1/10), common (≥1/100 to <1/10) or uncommon (≥1/1,000 to <1/100).
Table 2. Adverse drug reactions identified with tenofovir alafenamide:
| System organ class | |
|---|---|
| Frequency | Adverse reaction |
| Nervous system disorders | |
| Very common | Headache |
| Common | Dizziness |
| Gastrointestinal disorders | |
| Common | Diarrhoea, vomiting, nausea, abdominal pain, abdominal distension, flatulence |
| Hepatobiliary disorders | |
| Common | Increased ALT |
| Skin and subcutaneous tissue disorders | |
| Common | Rash, pruritus |
| Uncommon | Angioedema1, urticaria1 |
| Musculoskeletal and connective tissue disorders | |
| Common | Arthralgia |
| General disorders and administration site conditions | |
| Common | Fatigue |
1 Adverse reaction identified through post-marketing surveillance for tenofovir alafenamide-containing products.
In the open-label Phase 2 study (GS-US-320-4035; “Study 4035”) to evaluate the efficacy and safety of switching from another antiviral regimen to tenofovir alafenamide in virologically suppressed HBV infected patients, small median increases in fasting total cholesterol, direct low density lipoprotein (LDL), high density lipoprotein (HDL), and triglycerides from baseline to Week 96 were observed in patients with moderate or severe renal impairment (Part A Cohort 1) and patients with moderate or severe hepatic impairment (Part B), consistent with changes observed in Studies 108 and 110. Small median decreases in total cholesterol, LDL and triglycerides were observed in patients with ESRD on hemodialysis in Part A Cohort 2, while small median increases were observed in HDL from baseline to Week 96. Median (Q1, Q3) change from baseline at Week 96 in total cholesterol to HDL ratio was 0.1 (-0.4, 0.4) in the moderate or severe renal impairment group, and -0.4 (-0.8,-0.1) in patients with ESRD on hemodialysis and 0.1 (-0.2, 0.4) in patients with moderate or severe hepatic impairment.
Metabolic parameters
Body weight and levels of blood lipids and glucose may increase during therapy.
Other special populations
In Study 4035 in virologically suppressed patients with moderate to severe renal impairment (eGFR by Cockcroft-Gault method 15 to 59 mL/min; Part A, Cohort 1, N=78), end stage renal disease (ESRD) (eGFR <15 mL/min) on haemodialysis (Part A, Cohort 2, N=15), and/or moderate to severe hepatic impairment (Child-Pugh Class B or C at screening or by history; Part B, N=31) who switched from another antiviral regimen to tenofovir alafenamide, no additional adverse reactions to tenofovir alafenamide were identified through Week 96.
Paediatric population
The safety of tenofovir alafenamide was evaluated in 88 HBV-infected treatment-naïve and treatmentexperienced paediatric patients between the ages of 12 to <18 years weighing ≥35 kg (tenofovir alafenamide group N=47, placebo group N=23) and 6 to <12 years weighing ≥ 25 kg (tenofovir alafenamide group N=12, placebo group N=6) through Week 24 in a randomised, double-blind, placebo-controlled clinical study GS-US-320-1092 (“Study 1092”). After the double-blind phase, patients were switched to open-label tenofovir alafenamide at Week 24. The safety profile of tenofovir alafenamide in paediatric patients was comparable to that in adults. Reductions in bone mineral density (BMD ≥4%) of the lumbar spine and of whole body have been reported in some paediatric patients 6 years of age and older weighing at least 25 kg who received tenofovir alafenamide for up to 48 weeks (see sections 4.4 and 5.1).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.