VYTORIN Tablet Ref.[50754] Active ingredients: Ezetimibe Simvastatin
Source: FDA, National Drug Code (US) Revision Year: 2022
12.1. Mechanism of Action
VYTORIN
Plasma cholesterol is derived from intestinal absorption and endogenous synthesis. VYTORIN contains ezetimibe and simvastatin, two lipid-lowering compounds with complementary mechanisms of action. VYTORIN reduces elevated total-C, LDL-C, Apo B, TG, and non-HDL-C, and increases HDL-C through dual inhibition of cholesterol absorption and synthesis.
Ezetimibe
Ezetimibe reduces blood cholesterol by inhibiting the absorption of cholesterol by the small intestine. The molecular target of ezetimibe has been shown to be the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is involved in the intestinal uptake of cholesterol and phytosterols. In a 2-week clinical study in 18 hypercholesterolemic patients, ezetimibe inhibited intestinal cholesterol absorption by 54%, compared with placebo. Ezetimibe had no clinically meaningful effect on the plasma concentrations of the fat-soluble vitamins A, D, and E and did not impair adrenocortical steroid hormone production.
Ezetimibe localizes at the brush border of the small intestine and inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver. This causes a reduction of hepatic cholesterol stores and an increase in clearance of cholesterol from the blood; this distinct mechanism is complementary to that of statins [see Clinical Studies (14)].
Simvastatin
Simvastatin is a prodrug and is hydrolyzed to its active β-hydroxyacid form, simvastatin acid, after administration. Simvastatin is a specific inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the enzyme that catalyzes the conversion of HMG-CoA to mevalonate, an early and rate limiting step in the biosynthetic pathway for cholesterol. In addition, simvastatin reduces very-low-density lipoproteins (VLDL) and TG and increases HDL-C.
12.2. Pharmacodynamics
Clinical studies have demonstrated that elevated levels of total-C, LDL-C and Apo B, the major protein constituent of LDL, promote human atherosclerosis. In addition, decreased levels of HDL-C are associated with the development of atherosclerosis. Epidemiologic studies have established that cardiovascular morbidity and mortality vary directly with the level of total-C and LDL-C and inversely with the level of HDL-C. Like LDL, cholesterol-enriched triglyceride-rich lipoproteins, including VLDL, intermediate-density lipoproteins (IDL), and remnants, can also promote atherosclerosis. The independent effect of raising HDL-C or lowering TG on the risk of coronary and cardiovascular morbidity and mortality has not been determined.
12.3. Pharmacokinetics
The results of a bioequivalence study in healthy subjects demonstrated that the VYTORIN (ezetimibe and simvastatin) 10 mg/10 mg to 10 mg/80 mg combination tablets are bioequivalent to coadministration of corresponding doses of ezetimibe (ZETIA) and simvastatin (ZOCOR) as individual tablets.
Absorption
Ezetimibe
After oral administration, ezetimibe is absorbed and extensively conjugated to a pharmacologically active phenolic glucuronide (ezetimibe-glucuronide).
Simvastatin
The availability of the β-hydroxyacid to the systemic circulation following an oral dose of simvastatin was found to be less than 5% of the dose, consistent with extensive hepatic first-pass extraction.
Effect of Food on Oral Absorption
Ezetimibe
Concomitant food administration (high-fat or non-fat meals) had no effect on the extent of absorption of ezetimibe when administered as 10-mg tablets. The Cmax value of ezetimibe was increased by 38% with consumption of high-fat meals.
Simvastatin
Relative to the fasting state, the plasma profiles of both active and total inhibitors of HMG-CoA reductase were not affected when simvastatin was administered immediately before an American Heart Association recommended low-fat meal.
Distribution
Ezetimibe
Ezetimibe and ezetimibe-glucuronide are highly bound (>90%) to human plasma proteins.
Simvastatin
Both simvastatin and its β-hydroxyacid metabolite are highly bound (approximately 95%) to human plasma proteins. When radiolabeled simvastatin was administered to rats, simvastatin-derived radioactivity crossed the blood-brain barrier.
Metabolism and Excretion
Ezetimibe
Ezetimibe is primarily metabolized in the small intestine and liver via glucuronide conjugation with subsequent biliary and renal excretion. Minimal oxidative metabolism has been observed in all species evaluated.
In humans, ezetimibe is rapidly metabolized to ezetimibe-glucuronide. Ezetimibe and ezetimibe-glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10 to 20% and 80 to 90% of the total drug in plasma, respectively. Both ezetimibe and ezetimibe-glucuronide are eliminated from plasma with a half-life of approximately 22 hours for both ezetimibe and ezetimibe-glucuronide. Plasma concentration-time profiles exhibit multiple peaks, suggesting enterohepatic recycling.
Following oral administration of 14C-ezetimibe (20 mg) to human subjects, total ezetimibe (ezetimibe + ezetimibe-glucuronide) accounted for approximately 93% of the total radioactivity in plasma. After 48 hours, there were no detectable levels of radioactivity in the plasma.
Approximately 78% and 11% of the administered radioactivity were recovered in the feces and urine, respectively, over a 10-day collection period. Ezetimibe was the major component in feces and accounted for 69% of the administered dose, while ezetimibe-glucuronide was the major component in urine and accounted for 9% of the administered dose.
Simvastatin
Simvastatin is a lactone that is readily hydrolyzed in vivo to the corresponding β-hydroxyacid, a potent inhibitor of HMG-CoA reductase. Inhibition of HMG-CoA reductase is a basis for an assay in pharmacokinetic studies of the β-hydroxyacid metabolites (active inhibitors) and, following base hydrolysis, active plus latent inhibitors (total inhibitors) in plasma following administration of simvastatin. The major active metabolites of simvastatin present in human plasma are the β-hydroxyacid of simvastatin and its 6'-hydroxy, 6'-hydroxymethyl, and 6'-exomethylene derivatives.
Following an oral dose of 14C-labeled simvastatin in man, 13% of the dose was excreted in urine and 60% in feces. Plasma concentrations of total radioactivity (simvastatin plus 14C-metabolites) peaked at 4 hours and declined rapidly to about 10% of peak by 12 hours postdose.
Specific Populations
Geriatric Patients
Ezetimibe
In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were about 2-fold higher in older (≥65 years) healthy subjects compared to younger subjects.
Simvastatin
In a study including 16 elderly patients between 70 and 78 years of age who received simvastatin 40 mg/day, the mean plasma level of HMG-CoA reductase inhibitory activity was increased approximately 45% compared with 18 patients between 18-30 years of age.
Pediatric Patients
[See Use in Specific Populations (8.4)]
Gender
Ezetimibe
In a multiple-dose study with ezetimibe given 10 mg once daily for 10 days, plasma concentrations for total ezetimibe were slightly higher (<20%) in women than in men.
Race
Ezetimibe
Based on a meta-analysis of multiple-dose pharmacokinetic studies, there were no pharmacokinetic differences between Black and Caucasian subjects. Studies in Asian subjects indicated that the pharmacokinetics of ezetimibe was similar to those seen in Caucasian subjects.
Hepatic Impairment
Ezetimibe
After a single 10-mg dose of ezetimibe, the mean exposure (based on area under the curve [AUC]) to total ezetimibe was increased approximately 1.7-fold in patients with mild hepatic impairment (Child-Pugh score 5 to 6), compared to healthy subjects. The mean AUC values for total ezetimibe and ezetimibe increased approximately 3- to 4-fold and 5- to 6-fold, respectively, in patients with moderate (Child-Pugh score 7 to 9) or severe hepatic impairment (Child-Pugh score 10 to 15). In a 14-day, multiple-dose study (10 mg daily) in patients with moderate hepatic impairment, the mean AUC for total ezetimibe and ezetimibe increased approximately 4-fold compared to healthy subjects.
Renal Impairment
Ezetimibe
After a single 10-mg dose of ezetimibe in patients with severe renal disease (n=8; mean CrCl ≤30 mL/min/1.73 m 2), the mean AUC for total ezetimibe and ezetimibe increased approximately 1.5-fold, compared to healthy subjects (n=9).
Simvastatin
Pharmacokinetic studies with another statin having a similar principal route of elimination to that of simvastatin have suggested that for a given dose level higher systemic exposure may be achieved in patients with severe renal impairment (as measured by creatinine clearance).
Drug Interactions
[See also Drug Interactions (7)]
No clinically significant pharmacokinetic interaction was seen when ezetimibe was coadministered with simvastatin. No specific pharmacokinetic drug interaction studies with VYTORIN have been conducted other than the following study with NIASPAN (Niacin extended-release tablets).
Niacin: The effect of VYTORIN (10/20 mg daily for 7 days) on the pharmacokinetics of NIASPAN extended-release tablets (1000 mg for 2 days and 2000 mg for 5 days following a low-fat breakfast) was studied in healthy subjects. The mean Cmax and AUC of niacin increased 9% and 22%, respectively. The mean Cmax and AUC of nicotinuric acid increased 10% and 19%, respectively (N=13). In the same study, the effect of NIASPAN on the pharmacokinetics of VYTORIN was evaluated (N=15). While concomitant NIASPAN decreased the mean Cmax of total ezetimibe (1%), and simvastatin (2%), it increased the mean Cmax of simvastatin acid (18%). In addition, concomitant NIASPAN increased the mean AUC of total ezetimibe (26%), simvastatin (20%), and simvastatin acid (35%).
Cases of myopathy/rhabdomyolysis have been observed with simvastatin coadministered with lipid-modifying doses (≥1 g/day niacin) of niacin-containing products [See Warnings and Precautions (5.1) and Drug Interactions (7.4)].
Cytochrome P450: Ezetimibe had no significant effect on a series of probe drugs (caffeine, dextromethorphan, tolbutamide, and IV midazolam) known to be metabolized by cytochrome P450 (1A2, 2D6, 2C8/9 and 3A4) in a “cocktail” study of twelve healthy adult males. This indicates that ezetimibe is neither an inhibitor nor an inducer of these cytochrome P450 isozymes, and it is unlikely that ezetimibe will affect the metabolism of drugs that are metabolized by these enzymes.
In a study of 12 healthy volunteers, simvastatin at the 80-mg dose had no effect on the metabolism of the probe cytochrome P450 isoform 3A4 (CYP3A4) substrates midazolam and erythromycin. This indicates that simvastatin is not an inhibitor of CYP3A4 and, therefore, is not expected to affect the plasma levels of other drugs metabolized by CYP3A4.
Simvastatin acid is a substrate of the transport protein OATP1B1. Concomitant administration of medicinal products that are inhibitors of the transport protein OATP1B1 may lead to increased plasma concentrations of simvastatin acid and an increased risk of myopathy. For example, cyclosporine has been shown to increase the AUC of statins; although the mechanism is not fully understood, the increase in AUC for simvastatin acid is presumably due, in part, to inhibition of CYP3A4 and/or OATP1B1.
Simvastatin is a substrate for CYP3A4. Inhibitors of CYP3A4 can raise the plasma levels of HMG-CoA reductase inhibitory activity and increase the risk of myopathy. [See Warnings and Precautions (5.1); Drug Interactions (7.1).]
Ezetimibe
Table 4. Effect of Coadministered Drugs on Total Ezetimibe:
| Coadministered Drug and Dosing Regimen | Total Ezetimibe* | |
|---|---|---|
| Change in AUC | Change in Cmax | |
| Cyclosporine-stable dose required (75-150 mg BID)†,‡ | ↑240% | ↑290% |
| Fenofibrate, 200 mg QD, 14 days‡ | ↑48% | ↑64% |
| Gemfibrozil, 600 mg BID, 7 days‡ | ↑64% | ↑91% |
| Cholestyramine, 4 g BID, 14 days‡ | ↓55% | ↓4% |
| Aluminum & magnesium hydroxide combination antacid, single dose§ | ↓4% | ↓30% |
| Cimetidine, 400 mg BID, 7 days | ↑6% | ↑22% |
| Glipizide, 10 mg, single dose | ↑4% | ↓8% |
| Statins | ||
| Lovastatin 20 mg QD, 7 days | ↑9% | ↑3% |
| Pravastatin 20 mg QD, 14 days | ↑7% | ↑23% |
| Atorvastatin 10 mg QD, 14 days | ↓2% | ↑12% |
| Rosuvastatin 10 mg QD, 14 days | ↑13% | ↑18% |
| Fluvastatin 20 mg QD, 14 days | ↓19% | ↑7% |
* Based on 10 mg-dose of ezetimibe.
† Post-renal transplant patients with mild impaired or normal renal function. In a different study, a renal transplant patient with severe renal insufficiency (creatinine clearance of 13.2 mL/min/1.73 m ) who was receiving multiple medications, including cyclosporine, demonstrated a 12-fold greater exposure to total ezetimibe compared to healthy subjects.
‡ See 7. Drug Interactions.
§ Supralox, 20 mL.
Table 5. Effect of Ezetimibe Coadministration on Systemic Exposure to Other Drugs:
| Coadministered Drug and its Dosage Regimen | Ezetimibe Dosage Regimen | Change in AUC of Coadministered Drug | Change in Cmax of Coadministered Drug |
|---|---|---|---|
| Warfarin, 25 mg single dose on Day 7 | 10 mg QD, 11 days | ↓2% (R-warfarin) ↓4% (S-warfarin) | ↑3% (R-warfarin) ↑1% (S-warfarin) |
| Digoxin, 0.5 mg single dose | 10 mg QD, 8 days | ↑2% | ↓7% |
| Gemfibrozil, 600 mg BID, 7 days* | 10 mg QD, 7 days | ↓1% | ↓11% |
| Ethinyl estradiol & Levonorgestrel, QD, 21 days | 10 mg QD, Days 8-14 of 21 day oral contraceptive cycle | Ethinyl estradiol 0% Levonorgestrel 0% | Ethinyl estradiol ↓9% Levonorgestrel ↓5% |
| Glipizide, 10 mg on Days 1 and 9 | 10 mg QD, Days 2-9 | ↓3% | ↓5% |
| Fenofibrate, 200 mg QD, 14 days* | 10 mg QD, 14 days | ↑11% | ↑7% |
| Cyclosporine, 100 mg single dose Day 7* | 20 mg QD, 8 days | ↑15% | ↑10% |
| Statins | |||
| Lovastatin 20 mg QD, 7 days | 10 mg QD, 7 days | ↑19% | ↑3% |
| Pravastatin 20 mg QD, 14 days | 10 mg QD, 14 days | ↓20% | ↓24% |
| Atorvastatin 10 mg QD, 14 days | 10 mg QD, 14 days | ↓4% | ↑7% |
| Rosuvastatin 10 mg QD, 14 days | 10 mg QD, 14 days | ↑19% | ↑17% |
| Fluvastatin 20 mg QD, 14 days | 10 mg QD, 14 days | ↓39% | ↓27% |
Simvastatin
Table 6. Effect of Coadministered Drugs or Grapefruit Juice on Simvastatin Systemic Exposure:
| Coadministered Drug or Grapefruit Juice | Dosing of Coadministered Drug or Grapefruit Juice | Dosing of Simvastatin | Geometric Mean Ratio (Ratio* with / without coadministered drug) No Effect = 1.00 | ||
|---|---|---|---|---|---|
| AUC | Cmax | ||||
| Contraindicated with VYTORIN [see Contraindications (4) and Warnings and Precautions (5.1)] | |||||
| Telithromycin† | 200 mg QD for 4 days | 80 mg | simvastatin acid‡ | 12 | 15 |
| simvastatin | 8.9 | 5.3 | |||
| Nelfinavir† | 1250 mg BID for 14 days | 20 mg QD for 28 days | simvastatin acid‡ | ||
| simvastatin | 6 | 6.2 | |||
| Itraconazole† | 200 mg QD for 4 days | 80 mg | simvastatin acid‡ | 13.1 | |
| simvastatin | 13.1 | ||||
| Posaconazole | 100 mg (oral suspension) QD for 13 days | 40 mg | simvastatin acid‡ | 7.3 | 9.2 |
| simvastatin | 10.3 | 9.4 | |||
| 200 mg (oral suspension) QD for 13 days | 40 mg | simvastatin acid‡ | 8.5 | 9.5 | |
| simvastatin | 10.6 | 11.4 | |||
| Gemfibrozil | 600 mg BID for 3 days | 40 mg | simvastatin acid‡ | 2.85 | 2.18 |
| simvastatin | 1.35 | 0.91 | |||
| Avoid grapefruit juice with VYTORIN [see Warnings and Precautions (5.1)] | |||||
| Grapefruit Juice§ (high dose) | 200 mL of double-strength TID¶ | 60 mg single dose | simvastatin acid | 7 | |
| simvastatin | 16 | ||||
| Grapefruit Juice§ (low dose) | 8 oz (about 237 mL) of single- strength# | 20 mg single dose | simvastatin acid | 1.3 | |
| simvastatin | 1.9 | ||||
| Avoid taking with >10/10 mg VYTORIN, based on clinical and/or postmarketing simvastatin experience [see Warnings and Precautions (5.1)] | |||||
| Verapamil SR | 240 mg QD Days 1-7 then 240 mg BID on Days 8-10 | 80 mg on Day 10 | simvastatin acid | 2.3 | 2.4 |
| simvastatin | 2.5 | 2.1 | |||
| Diltiazem | 120 mg BID for 10 days | 80 mg on Day 10 | simvastatin acid | 2.69 | 2.69 |
| simvastatin | 3.10 | 2.88 | |||
| Diltiazem | 120 mg BID for 14 days | 20 mg on Day 14 | simvastatin | 4.6 | 3.6 |
| Dronedarone | 400 mg BID for 14 days | 40 mg QD for 14 days | simvastatin acid | 1.96 | 2.14 |
| simvastatin | 3.90 | 3.75 | |||
| Avoid taking with >10/20 mg VYTORIN, based on clinical and/or postmarketing simvastatin experience [see Warnings and Precautions (5.1)] | |||||
| Amiodarone | 400 mg QD for 3 days | 40 mg on Day 3 | simvastatin acid | 1.75 | 1.72 |
| simvastatin | 1.76 | 1.79 | |||
| Amlodipine | 10 mg QD for 10 days | 80 mg on Day 10 | simvastatin acid | 1.58 | 1.56 |
| simvastatin | 1.77 | 1.47 | |||
| Ranolazine SR | 1000 mg BID for 7 days | 80 mg on Day 1 and Days 6-9 | simvastatin acid | 2.26 | 2.28 |
| simvastatin | 1.86 | 1.75 | |||
| Avoid taking with >10/20 mg VYTORIN (or 10/40 mg for patients who have previously taken 80 mg simvastatin chronically, e.g., for 12 months or more, without evidence of muscle toxicity), based on clinical experience | |||||
| Lomitapide | 60 mg QD for 7 days | 40 mg single dose | simvastatin acid | 1.7 | 1.6 |
| simvastatin | 2 | 2 | |||
| Lomitapide | 10 mg QD for 7 days | 20 mg single dose | simvastatin acid | 1.4 | 1.4 |
| simvastatin | 1.6 | 1.7 | |||
| No dosing adjustments required for the following: | |||||
| Fenofibrate | 160 mg QD for 14 days | 80 mg QD on Days 8-14 | simvastatin acid | 0.64 | 0.89 |
| simvastatin | 0.89 | 0.83 | |||
| Propranolol | 80 mg single dose | 80 mg single dose | total inhibitor | 0.79 | ↓ from 33.6 to 21.1 ng∙eq/mL |
| active inhibitor | 0.79 | ↓ from 7.0 to 4.7 ng∙eq/mL | |||
* Results based on a chemical assay except results with propranolol as indicated.
† Results could be representative of the following CYP3A4 inhibitors: ketoconazole, erythromycin, clarithromycin, HIV protease inhibitors, and nefazodone.
‡ Simvastatin acid refers to the β-hydroxyacid of simvastatin.
§ The effect of amounts of grapefruit juice between those used in these two studies on simvastatin pharmacokinetics has not been studied.
¶ Double-strength: one can of frozen concentrate diluted with one can of water. #Grapefruit juice was administered TID for 2 days, and 200 mL together with single dose simvastatin and 30 and 90 minutes following single dose simvastatin on Day 3.
# Single-strength: one can of frozen concentrate diluted with 3 cans of water. Grapefruit juice was administered with breakfast for 3 days, and simvastatin was administered in the evening on Day 3.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
VYTORIN
No animal carcinogenicity or fertility studies have been conducted with the combination of ezetimibe and simvastatin. The combination of ezetimibe with simvastatin did not show evidence of mutagenicity in vitro in a microbial mutagenicity (Ames) test with Salmonella typhimurium and Escherichia coli with or without metabolic activation. No evidence of clastogenicity was observed in vitro in a chromosomal aberration assay in human peripheral blood lymphocytes with ezetimibe and simvastatin with or without metabolic activation. There was no evidence of genotoxicity at doses up to 600 mg/kg with the combination of ezetimibe and simvastatin (1:1) in the in vivo mouse micronucleus test.
Ezetimibe
A 104-week dietary carcinogenicity study with ezetimibe was conducted in rats at doses up to 1500 mg/kg/day (males) and 500 mg/kg/day (females) (~20 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe). A 104-week dietary carcinogenicity study with ezetimibe was also conducted in mice at doses up to 500 mg/kg/day (>150 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe). There were no statistically significant increases in tumor incidences in drug-treated rats or mice.
No evidence of mutagenicity was observed in vitro in a microbial mutagenicity (Ames) test with Salmonella typhimurium and Escherichia coli with or without metabolic activation. No evidence of clastogenicity was observed in vitro in a chromosomal aberration assay in human peripheral blood lymphocytes with or without metabolic activation. In addition, there was no evidence of genotoxicity in the in vivo mouse micronucleus test.
In oral (gavage) fertility studies of ezetimibe conducted in rats, there was no evidence of reproductive toxicity at doses up to 1000 mg/kg/day in male or female rats (~7 times the human exposure at 10 mg daily based on AUC0-24hr for total ezetimibe).
Simvastatin
In a 72-week carcinogenicity study, mice were administered daily doses of simvastatin of 25, 100, and 400 mg/kg body weight, which resulted in mean plasma drug levels approximately 1, 4, and 8 times higher than the mean human plasma drug level, respectively, (as total inhibitory activity based on AUC) after an 80-mg oral dose. Liver carcinomas were significantly increased in high-dose females and mid- and high-dose males with a maximum incidence of 90% in males. The incidence of adenomas of the liver was significantly increased in mid- and high-dose females. Drug treatment also significantly increased the incidence of lung adenomas in mid- and high-dose males and females. Adenomas of the Harderian gland (a gland of the eye of rodents) were significantly higher in high-dose mice than in controls. No evidence of a tumorigenic effect was observed at 25 mg/kg/day.
In a separate 92-week carcinogenicity study in mice at doses up to 25 mg/kg/day, no evidence of a tumorigenic effect was observed (mean plasma drug levels were 1 times higher than humans given 80 mg simvastatin as measured by AUC).
In a two-year study in rats at 25 mg/kg/day, there was a statistically significant increase in the incidence of thyroid follicular adenomas in female rats exposed to approximately 11 times higher levels of simvastatin than in humans given 80 mg simvastatin (as measured by AUC).
A second two-year rat carcinogenicity study with doses of 50 and 100 mg/kg/day produced hepatocellular adenomas and carcinomas (in female rats at both doses and in males at 100 mg/kg/day). Thyroid follicular cell adenomas were increased in males and females at both doses; thyroid follicular cell carcinomas were increased in females at 100 mg/kg/day. The increased incidence of thyroid neoplasms appears to be consistent with findings from other statins. These treatment levels represented plasma drug levels (AUC) of approximately 7 and 15 times (males) and 22 and 25 times (females) the mean human plasma drug exposure after an 80-mg daily dose.
No evidence of mutagenicity was observed in a microbial mutagenicity (Ames) test with or without rat or mouse liver metabolic activation. In addition, no evidence of damage to genetic material was noted in an in vitro alkaline elution assay using rat hepatocytes, a V-79 mammalian cell forward mutation study, an in vitro chromosome aberration study in CHO cells, or an in vivo chromosomal aberration assay in mouse bone marrow.
There was decreased fertility in male rats treated with simvastatin for 34 weeks at 25 mg/kg body weight (4 times the maximum human exposure level, based on AUC, in patients receiving 80 mg/day); however, this effect was not observed during a subsequent fertility study in which simvastatin was administered at this same dose level to male rats for 11 weeks (the entire cycle of spermatogenesis including epididymal maturation). No microscopic changes were observed in the testes of rats from either study. At 180 mg/kg/day (which produces exposure levels 22 times higher than those in humans taking 80 mg/day based on surface area, mg/m²), seminiferous tubule degeneration (necrosis and loss of spermatogenic epithelium) was observed. In dogs, there was drug-related testicular atrophy, decreased spermatogenesis, spermatocytic degeneration and giant cell formation at 10 mg/kg/day (approximately 2 times the human exposure, based on AUC, at 80 mg/day). The clinical significance of these findings is unclear.
13.2. Animal Toxicology and/or Pharmacology
CNS Toxicity
Optic nerve degeneration was seen in clinically normal dogs treated with simvastatin for 14 weeks at 180 mg/kg/day, a dose that produced mean plasma drug levels about 12 times higher than the mean plasma drug level in humans taking 80 mg/day.
A chemically similar drug in this class also produced optic nerve degeneration (Wallerian degeneration of retinogeniculate fibers) in clinically normal dogs in a dose-dependent fashion starting at 60 mg/kg/day, a dose that produced mean plasma drug levels about 30 times higher than the mean plasma drug level in humans taking the highest recommended dose (as measured by total enzyme inhibitory activity). This same drug also produced vestibulocochlear Wallerian-like degeneration and retinal ganglion cell chromatolysis in dogs treated for 14 weeks at 180 mg/kg/day, a dose that resulted in a mean plasma drug level similar to that seen with the 60 mg/kg/day dose.
CNS vascular lesions, characterized by perivascular hemorrhage and edema, mononuclear cell infiltration of perivascular spaces, perivascular fibrin deposits and necrosis of small vessels, were seen in dogs treated with simvastatin at a dose of 360 mg/kg/day, a dose that produced mean plasma drug levels that were about 14 times higher than the mean plasma drug levels in humans taking 80 mg/day. Similar CNS vascular lesions have been observed with several other drugs of this class.
There were cataracts in female rats after two years of treatment with 50 and 100 mg/kg/day (22 and 25 times the human AUC at 80 mg/day, respectively) and in dogs after three months at 90 mg/kg/day (19 times) and at two years at 50 mg/kg/day (5 times).
Ezetimibe
The hypocholesterolemic effect of ezetimibe was evaluated in cholesterol-fed Rhesus monkeys, dogs, rats, and mouse models of human cholesterol metabolism. Ezetimibe was found to have an ED50 value of 0.5 µg/kg/day for inhibiting the rise in plasma cholesterol levels in monkeys. The ED50 values in dogs, rats, and mice were 7, 30, and 700 µg/kg/day, respectively. These results are consistent with ezetimibe being a potent cholesterol absorption inhibitor.
In a rat model, where the glucuronide metabolite of ezetimibe (ezetimibe-glucuronide) was administered intraduodenally, the metabolite was as potent as ezetimibe in inhibiting the absorption of cholesterol, suggesting that the glucuronide metabolite had activity similar to the parent drug.
In 1-month studies in dogs given ezetimibe (0.03 to 300 mg/kg/day), the concentration of cholesterol in gallbladder bile increased ~2- to 4-fold. However, a dose of 300 mg/kg/day administered to dogs for one year did not result in gallstone formation or any other adverse hepatobiliary effects. In a 14-day study in mice given ezetimibe (0.3 to 5 mg/kg/day) and fed a low-fat or cholesterol-rich diet, the concentration of cholesterol in gallbladder bile was either unaffected or reduced to normal levels, respectively.
A series of acute preclinical studies was performed to determine the selectivity of ezetimibe for inhibiting cholesterol absorption. Ezetimibe inhibited the absorption of 14C-cholesterol with no effect on the absorption of triglycerides, fatty acids, bile acids, progesterone, ethinyl estradiol, or the fat-soluble vitamins A and D.
In 4- to 12-week toxicity studies in mice, ezetimibe did not induce cytochrome P450 drug-metabolizing enzymes. In toxicity studies, a pharmacokinetic interaction of ezetimibe with statins (parents or their active hydroxy acid metabolites) was seen in rats, dogs, and rabbits.
14. Clinical Studies
14.1 Primary Hyperlipidemia
VYTORIN
VYTORIN reduces total-C, LDL-C, Apo B, TG, and non-HDL-C, and increases HDL-C in patients with hyperlipidemia. Maximal to near maximal response is generally achieved within 2 weeks and maintained during chronic therapy.
VYTORIN is effective in men and women with hyperlipidemia. Experience in non-Caucasians is limited and does not permit a precise estimate of the magnitude of the effects of VYTORIN.
Five multicenter, double-blind studies conducted with either VYTORIN or coadministered ezetimibe and simvastatin equivalent to VYTORIN in patients with primary hyperlipidemia are reported: two were comparisons with simvastatin, two were comparisons with atorvastatin, and one was a comparison with rosuvastatin.
In a multicenter, double-blind, placebo-controlled, 12-week trial, 1528 hyperlipidemic patients were randomized to one of ten treatment groups: placebo, ezetimibe (10 mg), simvastatin (10 mg, 20 mg, 40 mg, or 80 mg), or VYTORIN (10/10, 10/20, 10/40, or 10/80).
When patients receiving VYTORIN were compared to those receiving all doses of simvastatin, VYTORIN significantly lowered total-C, LDL-C, Apo B, TG, and non-HDL-C. The effects of VYTORIN on HDL-C were similar to the effects seen with simvastatin. Further analysis showed VYTORIN significantly increased HDL-C compared with placebo. (See Table 7.) The lipid response to VYTORIN was similar in patients with TG levels greater than or less than 200 mg/dL.
Table 7. Response to VYTORIN in Patients with Primary Hyperlipidemia (Mean* % Change from Untreated Baseline†):
| Treatment | |||||||
|---|---|---|---|---|---|---|---|
| (Daily Dose) | N | Total-C | LDL-C | Apo B | HDL-C | TG* | Non-HDL-C |
| Pooled data (All VYTORIN doses)‡ | 609 | -38 | -53 | -42 | +7 | -24 | -49 |
| Pooled data (All simvastatin doses)‡ | 622 | -28 | -39 | -32 | +7 | -21 | -36 |
| Ezetimibe 10 mg | 149 | -13 | -19 | -15 | +5 | -11 | -18 |
| Placebo | 148 | -1 | -2 | 0 | 0 | -2 | -2 |
| VYTORIN by dose | |||||||
| 10/10 | 152 | -31 | -45 | -35 | +8 | -23 | -41 |
| 10/20 | 156 | -36 | -52 | -41 | +10 | -24 | -47 |
| 10/40 | 147 | -39 | -55 | -44 | +6 | -23 | -51 |
| 10/80 | 154 | -43 | -60 | -49 | +6 | -31 | -56 |
| Simvastatin by dose | |||||||
| 10 mg | 158 | -23 | -33 | -26 | +5 | -17 | -30 |
| 20 mg | 150 | -24 | -34 | -28 | +7 | -18 | -32 |
| 40 mg | 156 | -29 | -41 | -33 | +8 | -21 | -38 |
| 80 mg | 158 | -35 | -49 | -39 | +7 | -27 | -45 |
* For triglycerides, median % change from baseline.
† Baseline – on no lipid-lowering drug.
‡ VYTORIN doses pooled (10/10-10/80) significantly reduced total-C, LDLC, Apo B, TG, and non-HDL-C compared to simvastatin and significantly increased HDL-C compared to placebo.
In a multicenter, double-blind, controlled, 23-week study, 710 patients with known CHD or CHD risk equivalents, as defined by the NCEP ATP III guidelines, and an LDL-C ≥130 mg/dL were randomized to one of four treatment groups: coadministered ezetimibe and simvastatin equivalent to VYTORIN (10/10, 10/20, and 10/40) or simvastatin 20 mg. Patients not reaching an LDL-C <100 mg/dL had their simvastatin dose titrated at 6-week intervals to a maximal dose of 80 mg.
At Week 5, the LDL-C reductions with VYTORIN 10/10, 10/20, or 10/40 were significantly larger than with simvastatin 20 mg (see Table 8).
Table 8. Response to VYTORIN after 5 Weeks in Patients with CHD or CHD Risk Equivalents and an LDL-C ≥130 mg/dL:
| Simvastatin 20 mg | VYTORIN 10/10 | VYTORIN 10/20 | VYTORIN 10/40 | |
|---|---|---|---|---|
| N | 253 | 251 | 109 | 97 |
| Mean baseline LDL-C | 174 | 165 | 167 | 171 |
| Percent change LDL-C | -38 | -47 | -53 | -59 |
In a multicenter, double-blind, 6-week study, 1902 patients with primary hyperlipidemia, who had not met their NCEP ATP III target LDL-C goal, were randomized to one of eight treatment groups: VYTORIN (10/10, 10/20, 10/40, or 10/80) or atorvastatin (10 mg, 20 mg, 40 mg, or 80 mg).
Across the dosage range, when patients receiving VYTORIN were compared to those receiving milligram-equivalent statin doses of atorvastatin, VYTORIN lowered total-C, LDL-C, Apo B, and non-HDL-C significantly more than atorvastatin. Only the 10/40 mg and 10/80 mg VYTORIN doses increased HDL-C significantly more than the corresponding milligram-equivalent statin dose of atorvastatin. The effects of VYTORIN on TG were similar to the effects seen with atorvastatin. (See Table 9.)
Table 9. Response to VYTORIN and Atorvastatin in Patients with Primary Hyperlipidemia (Mean* % Change from Untreated Baseline†):
| Treatment | |||||||
|---|---|---|---|---|---|---|---|
| (Daily Dose) | N | Total-C‡ | LDL-C‡ | Apo B‡ | HDL-C | TG* | Non-HDL-C‡ |
| VYTORIN by dose | |||||||
| 10/10 | 230 | -34§ | -47§ | -37§ | +8 | -26 | -43§ |
| 10/20 | 233 | -37§ | -51§ | -40§ | +7 | -25 | -46§ |
| 10/40 | 236 | -41§ | -57§ | -46§ | +9§ | -27 | -52§ |
| 10/80 | 224 | -43§ | -59§ | -48§ | +8§ | -31 | -54§ |
| Atorvastatin by dose | |||||||
| 10 mg | 235 | -27 | -36 | -31 | +7 | -21 | -34 |
| 20 mg | 230 | -32 | -44 | -37 | +5 | -25 | -41 |
| 40 mg | 232 | -36 | -48 | -40 | +4 | -24 | -45 |
| 80 mg | 230 | -40 | -53 | -44 | +1 | -32 | -50 |
* For triglycerides, median % change from baseline.
† Baseline – on no lipid-lowering drug.
‡ VYTORIN doses pooled (10/10-10/80) provided significantly greater reductions in total-C, LDL-C, Apo B, and non-HDL-C compared to atorvastatin doses pooled (10-80).
§ p<0.05 for difference with atorvastatin at equal mg doses of the simvastatin component.
In a multicenter, double-blind, 24-week, forced-titration study, 788 patients with primary hyperlipidemia, who had not met their NCEP ATP III target LDL-C goal, were randomized to receive coadministered ezetimibe and simvastatin equivalent to VYTORIN (10/10 and 10/20) or atorvastatin 10 mg. For all three treatment groups, the dose of the statin was titrated at 6-week intervals to 80 mg. At each pre-specified dose comparison, VYTORIN lowered LDL-C to a greater degree than atorvastatin (see Table 10).
Table 10. Response to VYTORIN and Atorvastatin in Patients with Primary Hyperlipidemia (Mean* % Change from Untreated Baseline†):
| Treatment | N | Total-C | LDL-C | Apo B | HDL-C | TG* | Non-HDL-C |
|---|---|---|---|---|---|---|---|
| Week 6 | |||||||
| Atorvastatin 10 mg‡ | 262 | -28 | -37 | -32 | +5 | -23 | -35 |
| VYTORIN 10/10§ | 263 | -34¶ | -46¶ | -38¶ | +8¶ | -26 | -43¶ |
| VYTORIN 10/20# | 263 | -36¶ | -50¶ | -41¶ | +10¶ | -25 | -46¶ |
| Week 12 | |||||||
| Atorvastatin 20 mg | 246 | -33 | -44 | -38 | +7 | -28 | -42 |
| VYTORIN 10/20 | 250 | -37¶ | -50¶ | -41¶ | +9 | -28 | -46¶ |
| VYTORIN 10/40 | 252 | -39¶ | -54¶ | -45¶ | +12¶ | -31 | -50¶ |
| Week 18 | |||||||
| Atorvastatin 40 mg | 237 | -37 | -49 | -42 | +8 | -31 | -47 |
| VYTORIN 10/40Þ | 482 | -40¶ | -56¶ | -45¶ | +11¶ | -32 | -52¶ |
| Week 24 | |||||||
| Atorvastatin 80 mg | 228 | -40 | -53 | -45 | +6 | -35 | -50 |
| VYTORIN 10/80Þ | 459 | -43¶ | -59¶ | -49¶ | +12¶ | -35 | -55¶ |
* For triglycerides, median % change from baseline.
† Baseline – on no lipid-lowering drug.
‡ Atorvastatin: 10 mg start dose titrated to 20 mg, 40 mg, and 80 mg through Weeks 6, 12, 18, and 24.
§ VYTORIN: 10/10 start dose titrated to 10/20, 10/40, and 10/80 through Weeks 6, 12, 18, and 24.
¶ p≤0.05 for difference with atorvastatin in the specified week.
# VYTORIN: 10/20 start dose titrated to 10/40, 10/40, and 10/80 through Weeks 6, 12, 18, and 24.
Þ Data pooled for common doses of VYTORIN at Weeks 18 and 24.
In a multicenter, double-blind, 6-week study, 2959 patients with primary hyperlipidemia, who had not met their NCEP ATP III target LDL-C goal, were randomized to one of six treatment groups: VYTORIN (10/20, 10/40, or 10/80) or rosuvastatin (10 mg, 20 mg, or 40 mg).
The effects of VYTORIN and rosuvastatin on total-C, LDL-C, Apo B, TG, non-HDL-C and HDL-C are shown in Table 11.
Table 11. Response to VYTORIN and Rosuvastatin in Patients with Primary Hyperlipidemia (Mean* % Change from Untreated Baseline†):
| Treatment (Daily Dose) | N | Total-C‡ | LDL-C‡ | Apo B‡ | HDL-C | TG* | Non-HDL-C‡ |
|---|---|---|---|---|---|---|---|
| VYTORIN by dose | |||||||
| 10/20 | 476 | -37§ | -52§ | -42§ | +7 | -23§ | -47§ |
| 10/40 | 477 | -39¶ | -55¶ | -44¶ | +8 | -27 | -50¶ |
| 10/80 | 474 | -44# | -61# | -50# | +8 | -30# | -56# |
| Rosuvastatin by dose | |||||||
| 10 mg | 475 | -32 | -46 | -37 | +7 | -20 | -42 |
| 20 mg | 478 | -37 | -52 | -43 | +8 | -26 | -48 |
| 40 mg | 475 | -41 | -57 | -47 | +8 | -28 | -52 |
* For triglycerides, median % change from baseline.
† Baseline – on no lipid-lowering drug.
‡ VYTORIN doses pooled (10/20-10/80) provided significantly greater reductions in total-C, LDL-C, Apo B, and non-HDL-C compared to rosuvastatin doses pooled (10-40 mg).
§ p<0.05 vs. rosuvastatin 10 mg.
¶ p<0.05 vs. rosuvastatin 20 mg.
# p<0.05 vs. rosuvastatin 40 mg.
In a multicenter, double-blind, 24-week trial, 214 patients with type 2 diabetes mellitus treated with thiazolidinediones (rosiglitazone or pioglitazone) for a minimum of 3 months and simvastatin 20 mg for a minimum of 6 weeks were randomized to receive either simvastatin 40 mg or the coadministered active ingredients equivalent to VYTORIN 10/20. The median LDL-C and HbA1c levels at baseline were 89 mg/dL and 7.1%, respectively.
VYTORIN 10/20 was significantly more effective than doubling the dose of simvastatin to 40 mg. The median percent changes from baseline for VYTORIN vs. simvastatin were: LDL-C -25% and -5%; total-C -16% and -5%; Apo B -19% and -5%; and non-HDL-C -23% and -5%. Results for HDL-C and TG between the two treatment groups were not significantly different.
Ezetimibe
In two multicenter, double-blind, placebo-controlled, 12-week studies in 1719 patients with primary hyperlipidemia, ezetimibe significantly lowered total-C (-13%), LDL-C (-19%), Apo B (-14%), and TG (-8%), and increased HDL-C (+3%) compared to placebo. Reduction in LDL-C was consistent across age, sex, and baseline LDL-C.
Simvastatin
In two large, placebo-controlled clinical trials, the Scandinavian Simvastatin Survival Study (N=4,444 patients) and the Heart Protection Study (N=20,536 patients), the effects of treatment with simvastatin were assessed in patients at high risk of coronary events because of existing coronary heart disease, diabetes, peripheral vessel disease, history of stroke or other cerebrovascular disease. Simvastatin was proven to reduce: the risk of total mortality by reducing CHD deaths; the risk of non-fatal myocardial infarction and stroke; and the need for coronary and non-coronary revascularization procedures.
No incremental benefit of VYTORIN on cardiovascular morbidity and mortality over and above that demonstrated for simvastatin has been established.
14.2 Homozygous Familial Hypercholesterolemia (HoFH)
A double-blind, randomized, 12-week study was performed in patients with a clinical and/or genotypic diagnosis of HoFH. Data were analyzed from a subgroup of patients (n=14) receiving simvastatin 40 mg at baseline. Increasing the dose of simvastatin from 40 to 80 mg (n=5) produced a reduction of LDL-C of 13% from baseline on simvastatin 40 mg. Coadministered ezetimibe and simvastatin equivalent to VYTORIN (10/40 and 10/80 pooled, n=9), produced a reduction of LDL-C of 23% from baseline on simvastatin 40 mg. In those patients coadministered ezetimibe and simvastatin equivalent to VYTORIN (10/80, n=5), a reduction of LDL-C of 29% from baseline on simvastatin 40 mg was produced.
14.3 Chronic Kidney Disease (CKD)
The Study of Heart and Renal Protection (SHARP) was a multinational, randomized, placebo-controlled, double-blind trial that investigated the effect of VYTORIN on the time to a first major vascular event (MVE) among 9438 patients with moderate to severe chronic kidney disease (approximately one-third on dialysis at baseline) who did not have a history of myocardial infarction or coronary revascularization. An MVE was defined as nonfatal MI, cardiac death, stroke, or any revascularization procedure. Patients were allocated to treatment using a method that took into account the distribution of 8 important baseline characteristics of patients already enrolled and minimized the imbalance of those characteristics across the groups.
For the first year, 9438 patients were allocated 4:4:1, to VYTORIN 10/20, placebo, or simvastatin 20 mg daily, respectively. The 1-year simvastatin arm enabled the comparison of VYTORIN to simvastatin with regard to safety and effect on lipid levels. At 1 year the simvastatin-only arm was re-allocated 1:1 to VYTORIN 10/20 or placebo. A total of 9270 patients were ever allocated to VYTORIN 10/20 (n=4650) or placebo (n=4620) during the trial. The median follow-up duration was 4.9 years. Patients had a mean age of 61 years; 63% were male, 72% were Caucasian, and 23% were diabetic; and, for those not on dialysis at baseline, the median serum creatinine was 2.5 mg/dL and the median estimated glomerular filtration rate (eGFR) was 25.6 mL/min/1.73 m 2, with 94% of patients having an eGFR < 45 mL/min/1.73m 2. Eligibility did not depend on lipid levels. Mean LDL-C at baseline was 108 mg/dL. At 1 year, the mean LDL-C was 26% lower in the simvastatin arm and 38% lower in the VYTORIN arm relative to placebo. At the midpoint of the study (2.5 years), the mean LDL-C was 32% lower for VYTORIN relative to placebo. Patients no longer taking study medication were included in all lipid measurements.
In the primary intent-to-treat analysis, 639 (15.2%) of 4193 patients initially allocated to VYTORIN and 749 (17.9%) of 4191 patients initially allocated to placebo experienced an MVE. This corresponded to a relative risk reduction of 16% (p=0.001) (see Figure 1). Similarly, 526 (11.3%) of 4650 patients ever allocated to VYTORIN and 619 (13.4%) of 4620 patients ever allocated to placebo experienced a major atherosclerotic event (MAE; a subset of the MVE composite that excluded non-coronary cardiac deaths and hemorrhagic stroke), corresponding to a relative risk reduction of 17% (p=0.002). The trial demonstrated that treatment with VYTORIN 10/20 mg versus placebo reduced the risk for MVE and MAE in this CKD population. The study design precluded drawing conclusions regarding the independent contribution of either ezetimibe or simvastatin to the observed effect.
The treatment effect of VYTORIN on MVE was attenuated among patients on dialysis at baseline compared with those not on dialysis at baseline. Among 3023 patients on dialysis at baseline, VYTORIN reduced the risk of MVE by 6% (RR 0.94: 95% CI 0.80-1.09) compared with 22% (RR 0.78: 95% CI 0.69-0.89) among 6247 patients not on dialysis at baseline (interaction P=0.08).
Figure 1. Effect of VYTORIN on the Primary Endpoint of Risk of Major Vascular Events:
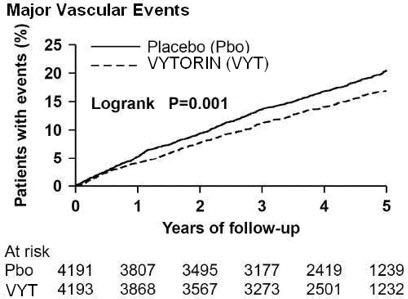
The individual components of MVE in all patients ever allocated to VYTORIN or placebo are presented in Table 12.
Table 12. Number of First Events for Each Component of the Major Vascular Event Composite Endpoint in SHARP*:
| Outcome | VYTORIN 10/20 (N=4650) | Placebo (N=4620) | Risk Ratio (95% CI) | P-value |
|---|---|---|---|---|
| Major Vascular Events | 701 (15.1%) | 814 (17.6%) | 0.85 (0.77-0.94) | 0.001 |
| Nonfatal MI | 134 (2.9%) | 159 (3.4%) | 0.84 (0.66-1.05) | 0.12 |
| Cardiac Death | 253 (5.4%) | 272 (5.9%) | 0.93 (0.78-1.10) | 0.38 |
| Any Stroke | 171 (3.7%) | 210 (4.5%) | 0.81 (0.66-0.99) | 0.038 |
| Non-hemorrhagic Stroke | 131 (2.8%) | 174 (3.8%) | 0.75 (0.60-0.94) | 0.011 |
| Hemorrhagic Stroke | 45 (1.0%) | 37 (0.8%) | 1.21 (0.78-1.86) | 0.40 |
| Any Revascularization | 284 (6.1%) | 352 (7.6%) | 0.79 (0.68-0.93) | 0.004 |
* Intention-to-treat analysis on all SHARP patients ever allocated to VYTORIN or placebo.
Among patients not on dialysis at baseline, VYTORIN did not reduce the risk of progressing to end-stage renal disease compared with placebo (RR 0.97: 95% CI 0.89-1.05).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.