ZEJULA Hard capsule Ref.[7611] Active ingredients: Niraparib
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: GlaxoSmithKline (Ireland) Limited, 12 Riverwalk, Citywest Business Campus, Dublin 24, Ireland
Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, other antineoplastic agents
ATC code: L01XK02
Mechanism of action and pharmacodynamic effects
Niraparib is an inhibitor of poly(ADP-ribose) polymerase (PARP) enzymes, PARP-1 and PARP-2, which play a role in DNA repair. In vitro studies have shown that niraparib-induced cytotoxicity may involve inhibition of PARP enzymatic activity and increased formation of PARP-DNA complexes resulting in DNA damage, apoptosis and cell death. Increased niraparib-induced cytotoxicity was observed in tumour cell lines with or without deficiencies in the BReast CAncer (BRCA) 1 and 2 tumour suppressor genes. In orthotopic high-grade serous ovarian cancer patient-derived xenograft tumours (PDX) grown in mice, niraparib has been shown to reduce tumour growth in BRCA 1 and 2 mutant, BRCA wild-type but homologous recombination (HR) deficient, and in tumours that are BRCA wild-type and without detectable HR deficiency.
Clinical efficacy and safety
First-line ovarian cancer maintenance treatment
PRIMA was a Phase 3 double-blind, placebo-controlled trial in which patients (n=733) in complete or partial response to first-line platinum-based chemotherapy were randomised 2:1 to niraparib or matched placebo. PRIMA was initiated with a starting dose of 300 mg QD in 475 patients (whereof 317 was randomised to the niraparib arm vs 158 in the placebo arm) in continuous 28-day cycles. The starting dose in PRIMA was changed with Amendment 2 of the Protocol. From that point forward, patients with a baseline body weight ≥77 kg and baseline platelet count ≥150,000/µL were administered niraparib 300 mg (n=34) or placebo daily (n=21) while patients with a baseline body weight <77 kg or baseline platelet count <150,000/μL were administered niraparib 200 mg (n=122) or placebo daily (n=61).
Patients were randomised post completion of first-line platinum-based chemotherapy plus/minus surgery. Subjects were randomised within 12 weeks of the first day of the last cycle of chemotherapy. Subjects had ≥6 and ≤9 cycles of platinum-based therapy. Following interval debulking surgery subjects had ≥2 post-operative cycles of platinum-based therapy. Patients who had received bevacizumab with chemotherapy but could not receive bevacizumab as maintenance therapy were not excluded from the study. Patients could not have received prior PARP inhibitor (PARPi) therapy, including niraparib. Patients who had neoadjuvant chemotherapy followed by interval debulking surgery could have visible residual or no residual disease. Patients with Stage III disease who had complete cytoreduction (i.e., no visible residual disease) after primary debulking surgery were excluded. Randomisation was stratified by best response during the front-line platinum regimen (complete response vs partial response), neoadjuvant chemotherapy (NACT) (Yes vs No); and homologous recombination deficiency (HRD) status [positive (HR deficient) vs negative (HR proficient) or not determined]. Testing for HRD was performed using the HRD test on tumour tissue obtained at the time of initial diagnosis. The CA-125 levels should be in the normal range (or a CA125 decrease by >90%) during the patient’s front-line therapy, and be stable for at least 7 days.
Patients began treatment on Cycle 1/Day 1 (C1/D1) with niraparib 200 or 300 mg or matched placebo administered QD in continuous 28-day cycles. Clinic visits occurred each cycle (4 weeks ± 3 days).
The primary endpoint was progression-free survival (PFS), as determined by blinded independent central review (BICR) per RECIST, version 1.1. Overall survival (OS) was a key secondary objective. PFS testing was performed hierarchically: first in the HR deficient population, then in the overall population. The median age of 62 ranged from 32 to 85 years among patients randomised with niraparib and 33 to 88 years among patients randomised with placebo. Eighty-nine percent of all patients were white. Sixty-nine percent of patients randomised with niraparib and 71% of patients randomised with placebo had an ECOG of 0 at study baseline. In the overall population, 65% of patients had stage III disease and 35% had stage IV disease. In the overall population, the primary tumour site in most patients (≥80%) was the ovary; most patients (>90%) had tumours with serous histology. Sixty-seven percent of the patients received NACT. Sixty-nine percent of the patients had a complete response to the first-line platinum-based chemotherapy. A total of 6 niraparib patients had received bevacizumab as prior treatment for their ovarian cancer.
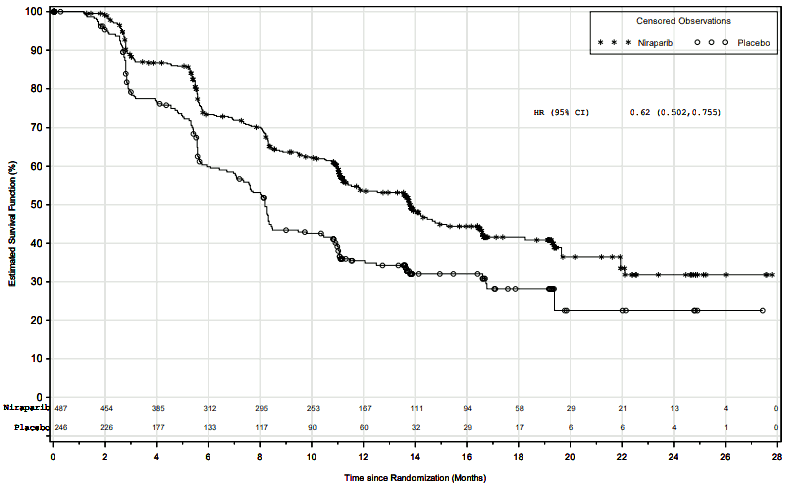
PRIMA demonstrated a statistically significant improvement in PFS for patients randomised to niraparib as compared with placebo in the HR deficient and overall population (Table 5, and Figures 1 and 2).
Secondary efficacy endpoints included PFS after the first subsequent therapy (PFS2) and OS (Table 5).
Table 5. Efficacy results – PRIMA (determined by BICR):
| HR deficient population | Overall population | |||
|---|---|---|---|---|
| niraparib (N=247) | placebo (N=126) | niraparib (N=487) | placebo (N=246) | |
| PFS median (95% CI) | 21.9 (19.3, NE) | 10.4 (8.1, 12.1) | 13.8 (11.5, 14.9) | 8.2 (7.3, 8.5) |
| Hazard ratio (95% CI) | 0.43 (0.31, 0.59) | 0.62 (0.50, 0.76) | ||
| p-value | <0.0001 | <0.0001 | ||
| PFS2 Hazard ratio (95% CI) | 0.84 (0.485, 1.453) | 0.81 (0.577, 1.139) | ||
| OS* Hazard ratio (95% CI) | 0.61 (0.265, 1.388) | 0.70 (0.44, 1.11) | ||
PFS = progression-free survival; CI = confidence interval; NE = not evaluable; OS = Overall survival; PFS2 = PFS after the first subsequent therapy.
* At the time of primary PFS analysis, an estimated survival at two years after randomization of 84% for patients receiving Zejula, as compared to 77% for patients receiving placebo in the overall population.
Data of PFS2 and OS are currently not mature.
Figure 1. Progression-free survival in patients with HR deficient tumours – PRIMA (ITT population, N=373):
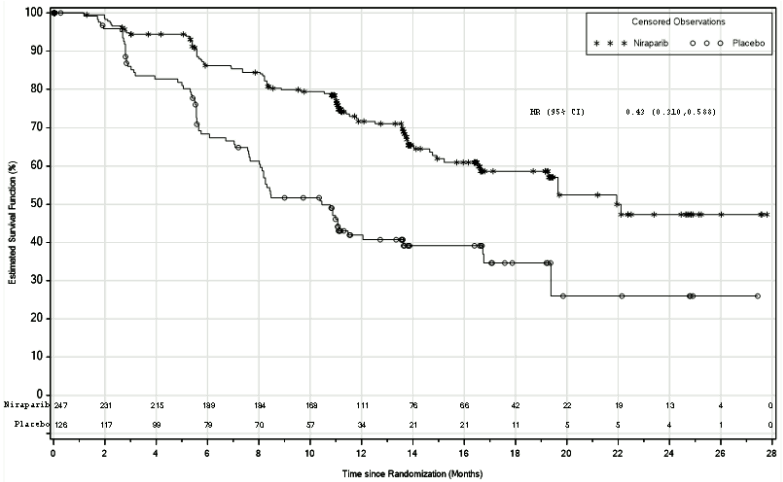
Figure 2. Progression-free survival in the overall population – PRIMA (ITT population, N=733):
Subgroup analyses
Within the HR deficient population, a hazard ratio of 0.40 (95% CI: 0.27, 0.62) was observed in the subgroup of patients with BRCAmut ovarian cancer (N=223). In the subgroup of HR deficient patients without a BRCA mutation (N=150), a hazard ratio of 0.50 (95% CI: 0.31, 0.83) was observed. In the HR proficient population (N=249), a hazard ratio of 0.68 (95% CI: 0.49, 0.94) was observed.
In exploratory subgroup analyses of patients who were administered 200 or 300 mg dose of Zejula based on baseline weight or platelet count, comparable efficacy (investigator-assessed PFS) was observed with a hazard ratio of 0.54 (95% CI: 0.33, 0.91) in the HR deficient population, and with a hazard ratio of 0.68 (95% CI: 0.49, 0.94) in the overall population. In the HR proficient subgroup, the dose of 200 mg appeared to give a lower treatment effect compared to the 300 mg dose.
Platinum-sensitive recurrent ovarian cancer maintenance treatment
The safety and efficacy of niraparib as maintenance therapy was studied in a Phase 3 randomised, double-blind, placebo-controlled international trial (NOVA) in patients with relapsed predominantly high grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer who were platinum sensitive, defined by complete response (CR) or partial response (PR) for more than six months to their penultimate (next to last) platinum-based therapy. To be eligible for niraparib treatment, the patient should be in response (CR or PR) following completion of last platinum-based chemotherapy. The CA-125 levels should be normal (or a >90% decrease in CA-125 from baseline) following their last platinum treatment, and be stable for at least 7 days. Patients could not have received prior PARPi therapy, including Zejula. Eligible patients were assigned to one of two cohorts based on the results of a germline BRCA (gBRCA) mutation test. Within each cohort, patients were randomised using a 2:1 allocation of niraparib and placebo. Patients were assigned to the gBRCAmut cohort based on blood samples for gBRCA analysis that were taken prior to randomisation. Testing for tumour BRCA (tBRCA) mutation and HRD was performed using the HRD test on tumour tissue obtained at the time of initial diagnosis or at the time of recurrence.
Randomisation within each cohort was stratified by time to progression after the penultimate platinum therapy before study enrolment (6 to <12 months and 12 months); use or not of bevacizumab in conjunction with the penultimate or last platinum regimen; and best response during the most recent platinum regimen (complete response and partial response).
Patients began treatment on Cycle 1/Day 1 (C1/D1) with niraparib 300 mg or matched placebo administered QD in continuous 28-day cycles. Clinic visits occurred each cycle (4 weeks ± 3 days).
In the NOVA study, 48% of patients had a dose interruption in Cycle 1. Approximately 47% of patients restarted at a reduced dose in Cycle 2.
The most commonly used dose in niraparib-treated patients in the NOVA study was 200 mg.
Progression-free survival (PFS) was determined per RECIST (Response Evaluation Criteria in Solid Tumors, version 1.1) or clinical signs and symptoms and increased CA-125. PFS was measured from the time of randomisation (which occurred up to 8 weeks after completion of the chemotherapy regimen) to disease progression or death.
The primary efficacy analysis for PFS was determined by blinded central independent assessment and was prospectively defined and assessed for the gBRCAmut cohort and the non-gBRCAmut cohort separately. Overall survival (OS) analyses were secondary outcome measures.
Secondary efficacy endpoints included chemotherapy-free interval (CFI), time to first subsequent therapy (TFST), PFS after the first subsequent therapy (PFS2), and OS.
Demographics, baseline disease characteristics, and prior treatment history were generally well balanced between the niraparib and placebo arms in the gBRCAmut (n=203) and the non-gBRCAmut cohorts (n=350). Median ages ranged from 57 to 63 years across treatments and cohorts. The primary tumour site in most patients (>80%) within each cohort was the ovary; most patients (>84%) had tumours with serous histology. A high proportion of patients in both treatment arms in both cohorts had received 3 or more prior lines of chemotherapy, including 49% and 34% of niraparib patients in the gBRCAmut and non-gBRCAmut cohorts, respectively. Most patients were age 18 to 64 years (78%), Caucasian (86%) and had an ECOG performance status of 0 (68%).
In the gBRCAmut cohort, the median number of treatment cycles was higher in the niraparib arm than the placebo arm (14 and 7 cycles, respectively). More patients in the niraparib group continued treatment for more than 12 months than patients in the placebo group (54.4% and 16.9% respectively).In the overall non-gBRCAmut cohort, the median number of treatment cycles was higher in the niraparib arm than in the placebo arm (8 and 5 cycles, respectively). More patients in the niraparib group continued treatment for more than 12 months than patients in the placebo group (34.2% and 21.1%, respectively).
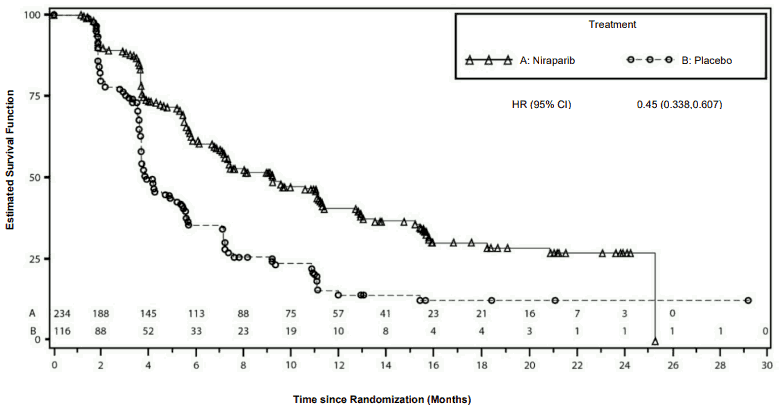
The study met its primary objective of statistically significantly improved PFS for niraparib maintenance monotherapy compared with placebo in the gBRCAmut cohort as well as in the overall non-gBRCAmut cohort. Table 6 and Figures 3 and 4 show the results for the PFS primary endpoint for the primary efficacy populations (gBRCAmut cohort and the overall non-gBRCAmut cohort).
Table 6. Summary of primary objective outcomes in the NOVA study:
| gBRCAmut cohort | Non-gBRCAmut cohort | |||
|---|---|---|---|---|
| niraparib (N=138) | placebo (N=65) | niraparib (N=234) | placebo (N=116) | |
| PFS median (95% CI) | 21.0 (12.9, NE) | 5.5 (3.8, 7.2) | 9.3 (7.2, 11.2) | 3.9 (3.7, 5.5) |
| p-value | <0.0001 | <0.0001 | ||
| Hazard ratio (Nir:plac) (95% CI) | 0.27 (0.173, 0.410) | 0.45 (0.338, 0.607) | ||
PFS = progression-free survival; CI = confidence interval; NE = not evaluable.
Figure 3. Kaplan-Meier plot for progression-free survival in the gBRCAmut cohort based on IRC assessment – NOVA (ITT population, N=203):
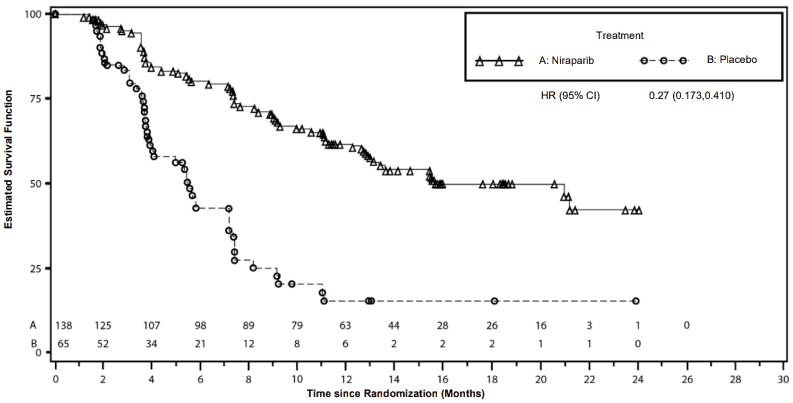
Figure 4. Kaplan-Meier plot for progression-free survival in the non-gBRCAmut cohort/overall based on IRC assessment – NOVA (ITT population, N=350):
Secondary efficacy endpoints in NOVA
At the final analysis, the median PFS2 in the gBRCAmut cohort was 29.9 months for patients treated with niraparib compared to 22.7 months for patients on placebo (HR=0.70; 95% CI: 0.50, 0.97). The median PFS2 in the non-gBRCAmut cohort was 19.5 months for patients treated with niraparib compared to 16.1 months for patients on placebo (HR=0.80; 95% CI: 0.63, 1.02).
At the final analysis of overall survival, the median OS in the gBRCAmut cohort (n=203) was 40.9 months for patients treated with niraparib compared with 38.1 months for patients on placebo (HR=0.85; 95% CI: 0.61, 1.20). The cohort maturity for the gBRCAmut cohort was 76%. The median OS in the non-gBRCAmut cohort (n=350) was 31.0 months for patients treated with niraparib compared with 34.8 months for patients on placebo (HR=1.06; 95% CI: 0.81, 1.37). The cohort maturity for the non-gBRCAmut cohort was 79%.
Patient-reported outcome (PRO) data from validated survey tools (FOSI and EQ-5D) indicate that niraparib-treated patients reported no difference from placebo in measures associated with quality of life (QoL).
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Zejula in all subsets of the paediatric population in ovarian carcinoma (excluding rhabdomyosarcoma and germ cell tumours).
Pharmacokinetic properties
Absorption
Following a single-dose administration of 300 mg niraparib under fasting conditions, niraparib was measurable in plasma within 30 minutes and the mean peak plasma concentration (Cmax) for niraparib was reached in about 3 hours [804 ng/mL (% CV: 50.2%)]. Following multiple oral doses of niraparib from 30 mg to 400 mg once daily, accumulation of niraparib was approximately 2 to 3 folds.
The systemic exposures (Cmax and AUC) to niraparib increased in a dose-proportional manner when the dose of niraparib increased from 30 mg to 400 mg. The absolute bioavailability of niraparib is approximately 73%, indicating minimal first pass effect. In a population pharmacokinetic analysis of niraparib, the inter-individual variability in bioavailability was estimated to a coefficient of variation (CV) of 31%.
A concomitant high-fat meal did not significantly affect the pharmacokinetics of niraparib after administration of 300 mg of niraparib. The tablet and capsule formulations have been demonstrated to be bioequivalent. Following administration of either one 300 mg tablet or three 100 mg capsules of niraparib in 108 patients with solid tumours under fasting conditions, the 90% confidence intervals of the geometric mean ratios for tablet compared to capsules for Cmax, AUClast and AUC∞ fell within the limits of bioequivalence (0.80 and 1.25).
Distribution
Niraparib was moderately protein bound in human plasma (83%), mainly with serum albumin. In a population pharmacokinetic analysis of niraparib, the apparent volume of distribution (Vd/F) was 1,311 L (based on a 70 kg patient) in cancer patients (CV 116%), indicating extensive tissue distribution of niraparib.
Biotransformation
Niraparib is metabolised primarily by carboxylesterases (CEs) to form a major inactive metabolite, M1. In a mass balance study, M1 and M10 (the subsequently formed M1 glucuronides) were the major circulating metabolites.
Elimination
Following a single oral 300-mg dose of niraparib, the mean terminal half-life (t½) of niraparib ranged from 48 to 51 hours (approximately 2 days). In a population pharmacokinetic analysis, the apparent total clearance (CL/F) of niraparib was 16.5 L/h in cancer patients (CV 23.4%).
Niraparib is eliminated primarily through the hepatobiliary and renal routes. Following an oral administration of a single 300-mg dose of [14C]-niraparib, on average 86.2% (range 71% to 91%) of the dose was recovered in urine and faeces over 21 days. Radioactive recovery in the urine accounted for 47.5% (range 33.4% to 60.2%) and in the faeces for 38.8% (range 28.3% to 47%) of the dose. In pooled samples collected over 6 days, 40% of the dose was recovered in the urine primarily as metabolites and 31.6% of the dose was recovered in the faeces primarily as unchanged niraparib.
Special populations
Renal impairment
In the population pharmacokinetic analysis, patients with mild (creatinine clearance 60-90 mL/min) and moderate (30-60 mL/min) renal impairment had mildly reduced niraparib clearance compared to individuals with normal renal function (7-17% higher exposure in mild and 17-38% higher exposure in moderate renal impairment). The difference in exposure is not considered to warrant dose adjustment. No patients with pre-existing severe renal impairment or end-stage renal disease undergoing hemodialysis were identified in clinical studies (see section 4.2).
Hepatic impairment
In the population pharmacokinetic analysis of data from clinical studies in patients, pre-existing mild hepatic impairment (n=155) did not influence the clearance of niraparib. In a clinical study of cancer patients using NCI-ODWG criteria to classify the degree of hepatic impairment, niraparib AUCinf in patients with moderate hepatic impairment (n=8) was 1.56 (90% CI: 1.06, 2.30) times the niraparib AUCinf in patients with normal hepatic function (n=9) following administration of a single 300 mg dose. Niraparib dose adjustment is recommended for patients with moderate hepatic impairment (see section 4.2). Moderate hepatic impairment did not have an effect on niraparib Cmax or on niraparib protein binding. The pharmacokinetics of niraparib have not been assessed in patients with severe hepatic impairment (see sections 4.2 and 4.4).
Weight, age and race
Increasing weight was found to increase niraparib volume of distribution in the population pharmacokinetic analysis. No impact of weight was identified on niraparib clearance or overall exposure. Dose adjustment according to body weight is not warranted from a pharmacokinetic point of view.
Increasing age was found to decrease niraparib clearance in the population pharmacokinetic analysis. The average exposure in a 91-year old patient was predicted to be 23% higher than in a 30-year old patient. The impact of age is not considered to warrant dose adjustment.
There is insufficient data across races to conclude on the impact of race on niraparib pharmacokinetics.
Paediatric population
No studies have been conducted to investigate the pharmacokinetics of niraparib in paediatric patients.
Preclinical safety data
Safety pharmacology
In vitro, niraparib inhibited the dopamine transporter DAT at concentration levels below human exposure levels. In mice, single doses of niraparib increased intracellular levels of dopamine and metabolites in cortex. Reduced locomotor activity was seen in one of two single dose studies in mice. The clinical relevance of these findings is not known. No effect on behavioural and/or neurological parameters have been observed in repeat-dose toxicity studies in rats and dogs at estimated CNS exposure levels similar to or below expected therapeutic exposure levels.
Repeat-dose toxicity
Decreased spermatogenesis was observed in rats and dogs at exposure levels below those seen clinically and was largely reversible within 4 weeks of cessation of dosing.
Genotoxicity
Niraparib was not mutagenic in a bacterial reverse mutation assay (Ames) test but was clastogenic in an in vitro mammalian chromosomal aberration assay and in an in vivo rat bone marrow micronucleus assay. This clastogenicity is consistent with genomic instability resulting from the primary pharmacology of niraparib and indicates potential for genotoxicity in humans.
Reproductive toxicology
Reproductive and developmental toxicity studies have not been conducted with niraparib.
Carcinogenicity
Carcinogenicity studies have not been conducted with niraparib.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.