Valbenazine
Chemical formula: C₂₄H₃₈N₂O₄ Molecular mass: 418.578 g/mol PubChem compound: 24795069
Mechanism of action
The mechanism of action of valbenazine for the treatment of tardive dyskinesia and chorea in patients with Huntington's disease is unclear, but is thought to be mediated through the reversible inhibition of vesicular monoamine transporter 2 (VMAT2), a transporter that regulates monoamine uptake from the cytoplasm to the synaptic vesicle for storage and release.
Pharmacodynamic properties
Valbenazine inhibits human VMAT2 (Ki ~150 nM) with no appreciable binding affinity for VMAT1 (Ki >10 µM). Valbenazine is converted to the active metabolite [+]-α-dihydrotetrabenazine ([ + ]-α-HTBZ).
[+]-α-HTBZ also binds with relatively high affinity to human VMAT2 (Ki ~3 nM).
Valbenazine and [+]-α-HTBZ have no appreciable binding affinity (Ki >5000 nM) for dopaminergic (including D2), serotonergic (including 5HT2B), adrenergic, histaminergic or muscarinic receptors.
Cardiac electrophysiology
Valbenazine may cause an increase in the corrected QT interval in patients who are CYP2D6 poor metabolizers or who are taking a strong CYP2D6 or CYP3A4 inhibitor. An exposure-response analysis of clinical data from two healthy volunteer studies revealed increased QTc interval with higher plasma concentrations of the active metabolite. Based on this model, patients taking an valbenazine 60 mg or 80 mg dose with increased exposure to the metabolite (e.g., being a CYP2D6 poor metabolizer) may have a mean (upper bound of double-sided 90% CI) QT prolongation of 9.6 (12.0) msec or 11.7 (14.7) msec, respectively as compared to otherwise healthy volunteers given valbenazine, who had a respective mean (upper bound of double-sided 90% CI) QT prolongation of 5.3 (6.7) msec or 6.7 (8.4) msec.
Pharmacokinetic properties
Valbenazine and its active metabolite ([+]-α-HTBZ) demonstrate approximate proportional increases for the area under the plasma concentration versus time curve (AUC) and maximum plasma concentration (Cmax) after single oral doses from 40 mg to 300 mg (i.e., 50% to 375% of the recommended treatment dose).
Absorption
Following oral administration of valbenazine, the time to reach maximum valbenazine plasma concentration (tmax) ranges from 0.5 to 1.0 hours. Valbenazine reaches steady state plasma concentrations within 1 week. The absolute oral bioavailability of valbenazine is approximately 49%. [+]-α-HTBZ gradually forms and reaches Cmax 4 to 8 hours after administration of valbenazine.
Following administration of 80 mg valbenazine orally as sprinkle on applesauce, the geometric mean peak plasma concentration (Cmax) of valbenazine was 512 ng/mL, area under the plasma concentration-time curve (AUCinf) was 5,600 ng*hr/mL and the median time to reach Cmax (Tmax) was 1.5 hours. For the [+]-α-HTBZ metabolite, the geometric mean Cmax was 23 ng/mL, AUCinf was 739 ng*hr/mL and the median Tmax was 6 hours.
Following oral administration of 80 mg valbenazine, the geometric mean Cmax, AUCinf and median Tmax of valbenazine were 744 ng/mL, 6,419 ng*hr/mL and 0.5 hour, respectively. For the [+]-α-HTBZ metabolite, the geometric mean Cmax was 26 ng/mL, AUCinf was 859 ng*hr/mL and the median Tmax was 6 hours.
When 80 mg valbenazine orally as sprinkle was swallowed with water, the geometric mean valbenazine Cmax, AUCinf and median Tmax were 685 ng/mL, 5,981 ng*hr/mL and 1 hour, respectively. For the [+]-α-HTBZ metabolite, the geometric mean Cmax was 23 ng/mL, AUCinf was 772 ng*hr/mL and the median Tmax was 6 hours.
Effect of food
Ingestion of a high-fat meal decreases valbenazine Cmax by approximately 47% and AUC by approximately 13%. [+]-α-HTBZ Cmax and AUC are unaffected.
Valbenazine orally as sprinkle:
Ingestion of a high-fat meal decreases valbenazine Cmax by approximately 15% and did not have an appreciable effect on AUC. [+]-α-HTBZ Cmax and AUC are unaffected.
Distribution
The plasma protein binding of valbenazine and [+]-α-HTBZ are greater than 99% and approximately 64%, respectively. The mean steady state volume of distribution of valbenazine is 92 L.
Nonclinical data in Long-Evans rats show that valbenazine can bind to melanin-containing structures of the eye such as the uveal tract. The relevance of this observation to clinical use of valbenazine is unknown.
Elimination
Valbenazine has a mean total plasma systemic clearance value of 7.2 L/hr. Valbenazine and [+]-α-HTBZ have half-lives of 15 to 22 hours.
Metabolism
Valbenazine is extensively metabolized after oral administration by hydrolysis of the valine ester to form the active metabolite ([ + ]-α-HTBZ) and by oxidative metabolism, primarily by CYP3A4/5, to form mono-oxidized valbenazine and other minor metabolites. [+]-α-HTBZ appears to be further metabolized in part by CYP2D6.
Excretion
Following the administration of a single 50-mg oral dose of radiolabeled C-valbenazine (i.e., ~63% of the recommended treatment dose), approximately 60% and 30% of the administered radioactivity was recovered in the urine and feces, respectively. Less than 2% was excreted as unchanged valbenazine or [+]-α-HTBZ in either urine or feces.
Specific populations
Exposures of valbenazine in patients with hepatic and severe renal impairment are summarized in Figure 1.
Figure 1. Effects of hepatic and severe renal impairment on valbenazine pharmacokinetics:
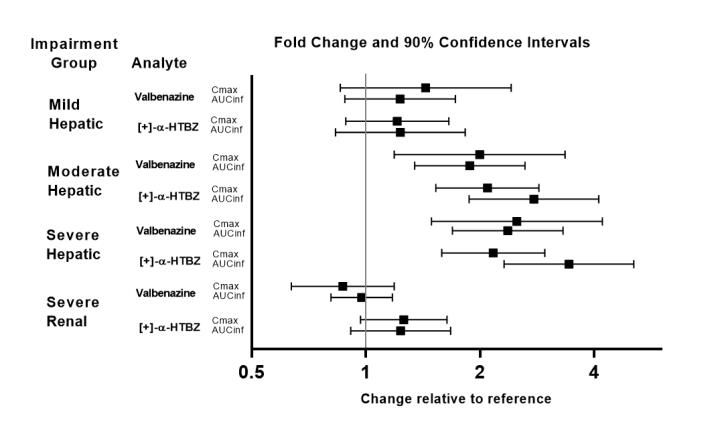
AUCinf=area under the plasma concentration versus time curve from 0 hours extrapolated to infinity
[ + ]-α-HTBZ=[+]-α-dihydrotetrabenazine (active metabolite)
Drug interaction studies
In vivo drug interactions
The effects of paroxetine, ketoconazole and rifampin on the exposure of valbenazine are summarized in Figure 2.
Figure 2. Effects of strong cyp2d6 and cyp3a4 inhibitors and cyp3a4 inducers on valbenazine pharmacokinetics:
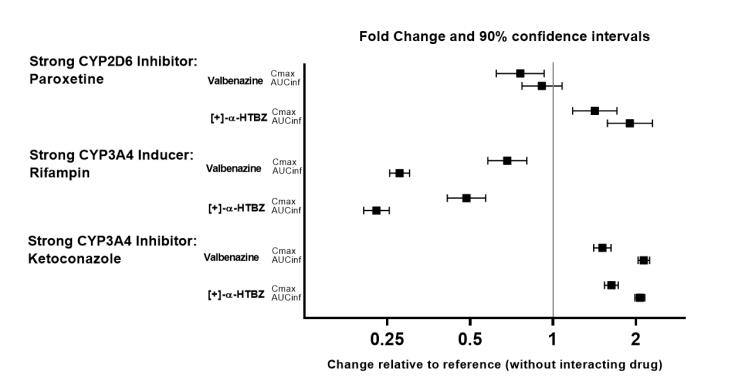
AUCinf=area under the plasma concentration versus time curve from 0 hours extrapolated to infinity
[ + ]-α-HTBZ=[+]-α-dihydrotetrabenazine (active metabolite)
The effects of valbenazine on the exposure of other coadministered drugs are summarized in Figure 3.
Figure 3. Effects of valbenazine on pharmacokinetics of other drugs:
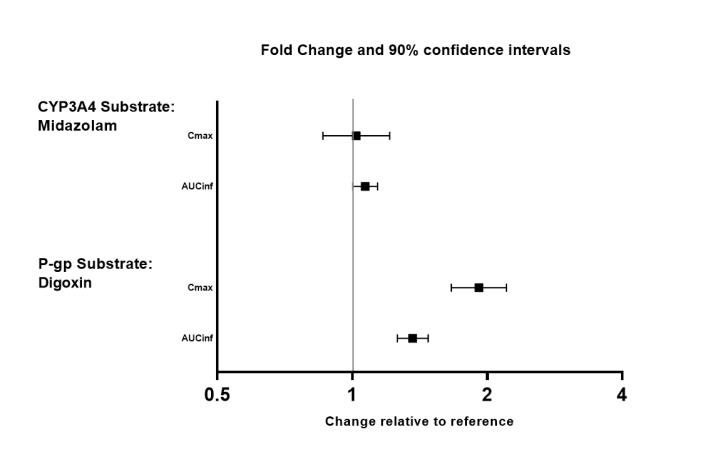
AUCinf=area under the plasma concentration versus time curve from 0 hours extrapolated to infinity
In vitro drug interactions
The results of in vitro studies suggest that valbenazine and [+]-α-HTBZ are unlikely to inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2E1 or CYP3A4/5, or induce CYP1A2, CYP2B6 or CYP3A4/5 at clinically relevant concentrations.
The results of in vitro studies suggest that valbenazine and [+]-α-HTBZ are unlikely to inhibit the transporters (BCRP, OAT1, OAT3, OCT2, OATP1B1, or OATP1B3) at clinically relevant concentrations.
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.