ATENEF Film-coated tablet Ref.[50537] Active ingredients: Efavirenz Emtricitabine Tenofovir disoproxil
Source: Health Products Regulatory Authority (ZA) Revision Year: 2022 Publisher: SONKE PHARMACEUTICALS (PTY) LTD, Ground Floor, Tugela House, Riverside Office Park, 1303 Heuwel Avenue, Centurion
4.3. Contraindications
- Hypersensitivity to efavirenz, emtricitabine, tenofovir disoproxil fumarate or to any of the excipients listed in section 6.1.
- ATENEF should not be administered concurrently with astemizole, bepridil, cisapride, midazolam, pimozide, triazolam or ergot derivatives because competition for CYP3A4 by efavirenz could result in inhibition of metabolism of these medicines and create the potential for serious and/or life-threatening adverse events (e.g. cardiac arrhythmias, prolonged sedation or respiratory depression). It should not be administeredconcurrently with voriconazole because efavirenz significantly decreases voriconazole plasma concentrations (see section 4.5).
- Pregnancy and lactation seesection 4.6).
- Moderate to severe renal impairment [Creatinine clearance less than 50 ml/min (see section 4.4 and section 5.2)].
- Patients with a history of previous liver injury/failure with efavirenz containing antiretroviral treatment (ART) (see section 4.4).
4.4. Special warnings and precautions for use
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, alone or in combination with other antiretrovirals.
ATENEF is not indicated for the treatment of chronic hepatitis B virus (HBV) infection and the safety and efficacy of ATENEF has not been established in patients co-infected with HBV and HIV. Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued emtricitabine or tenofovir, which are components of ATENEF. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV and HBV and discontinue ATENEF. If appropriate, initiation of anti-hepatitis B therapy may be warranted.
Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination with other antiretrovirals. A majority of these cases have been in women. Obesity and prolonged nucleoside exposure may be risk factors. Particular caution should be exercised when administering nucleoside analogues to any patient with known risk factors for liver disease; however, cases have also been reported in patients with no known risk factors. Treatment with ATENEF tablets should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
Routine testing of serum lactate levels in asymptomatic patients on ART is not recommended. Measurement of serum lactate levels is recommended only for patients presenting with clinical signs or symptoms consistent with lactic acidosis.
Lactate 2 to 5 mmol/l: monitor regularly and be alert for clinical signs.
Lactate 5 to 10 mmol/l without symptoms: monitor closely.
Lactate 5 to 10 mmol/l with symptoms: STOP all therapy. Exclude other causes (e.g. sepsis, uremia, diabetic ketoacidosis, thyrotoxicosis, lymphoma).
Lactate greater than or equal to 10 mmol/l: STOP all therapy (80% mortality in case studies).
Patients Co-infected with HIV-1 and HBV
It is recommended that all patients with HIV-1 be tested for the presence of chronic HBV before initiating antiretroviral therapy. ATENEF tablets are not indicated for the treatment of chronic HBV infection, and the safety and efficacy of ATENEF tablets have not been established in patients co-infected with HBV and HIV. Severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HBV and HIV and have discontinued emtricitabine or tenofovir DF. In some of these patients treated with emtricitabine, the exacerbations of hepatitis B were associated with liver decompensation and liver failure. Hepatic function should be monitored closely with both clinical and laboratory follow up for at least several months in patients who are co-infected with HIV and HBV and discontinue ATENEF tablets. If appropriate, initiation of anti-hepatitis B therapy may be warranted.
Co-administration with Related Medicines
Related medicines not for co-administration with ATENEF include emtricitabine, tenofovir DF, emtricitabine/tenofovir DF and efavirenz, which contain the same active components as ATENEF. Due to similarities between emtricitabine and lamivudine, ATENEF should not be co-administered with medicines containing lamivudine, including lamivudine/zidovudine, lamivudine, abacavir sulfate/lamivudine, or abacavir sulfate/lamivudine/zidovudine.
Medicine Interactions (see section 4.5)
Concomitant use of ATENEF and St. John’s wort (Hypericium perforatum) or St. John’s wort containing products is not recommended. Co-administration of NNRTIs, including efavirenz with St. John’s wort is expected to substantially decrease NNRTI concentrations and may result in suboptimal levels of efavirenz and may lead to virological response and possibly resistance to efavirenz or to the class of NNRTI’s.
Psychiatric Symptoms
Serious psychiatric adverse experiences have been reported in patients treated with efavirenz. These include: severe depression, suicidal ideation, nonfatal suicide attempts, aggressive behaviour, paranoid reactions, and manic reactions. Factors associated with an increase in the occurrence of these psychiatric symptoms are a history of injection medicine use, psychiatric history, and receipt of psychiatric medication. Cases of efavirenz-treated patients who discontinued or interrupted treatment because of one or more of these selected psychiatric symptoms have been reported. There have also been post-marketing reports of death by suicide, delusions, and psychosis-like behavior, although a causal relationship to the use of efavirenz cannot be determined from these reports. Patients with serious psychiatric adverse experiences should seek immediate medical evaluation to assess the possibility that the symptoms may be related to the use of efavirenz, and if so, to determine whether the risks of continued therapy outweigh the benefits (see section 4.8 ).
Nervous System Symptoms
These symptoms include dizziness, insomnia, impaired concentration, somnolence, abnormal dreams, and hallucinations. Other reported symptoms are euphoria, confusion, agitation, amnesia, stupor, abnormal thinking, and depersonalisation. Patients should be informed that these common symptoms are likely to improve with continued therapy and are not predictive of subsequent onset of the less frequent psychiatric symptoms (see section 4.4, Psychiatric Symptoms). Dosing at bedtime may improve the tolerability of these nervous system symptoms (see section 4.2 and section 4.6).
Analysis of long-term data from a study, (median follow-up 180 weeks, 102 weeks, and 76 weeks for patients treated with efavirenz + zidovudine + lamivudine, efavirenz + indinavir, and indinavir + zidovudine + lamivudine, respectively) showed that, beyond 24 weeks of therapy, the incidences of new-onset nervous system symptoms among efavirenz-treated subjects were generally similar to those in the indinavircontaining control arm.
Patients receiving ATENEF should be alerted to the potential for additive central nervous system effects when ATENEF are used concomitantly with alcohol or psychoactive medicines.
Renal Impairment (see section 4.3)
Emtricitabine and tenofovir are principally eliminated by the kidney; however, efavirenz is not. Since efavirenz, emtricitabine and tenofovir disoproxil fumarate fixed dose tablets is a combination product and the dose of the individual components cannot be altered, patients with creatinine clearance below 50 ml/min should not receive ATENEF.
eClcr (ml/min) = [140 – age] x Wt (kg) x 0.85 (if female) / Scr (μmol/L)
Renal impairment, including cases of acute renal failure and Fanconi syndrome (renal tubular injury with severe hypophosphataemia), has been reported in association with the use of tenofovir DF (see section 4.8).
It is recommended that creatinine clearance be calculated in all patients prior to initiating therapy and as clinically appropriate during therapy with efavirenz, emtricitabine and tenofovir disoproxil fumarate fixed dose combination tablets. Routine monitoring of calculated creatinine clearance and serum phosphorus should be performed in patients at risk for renal impairment. ATENEF should be avoided with concurrent or recent use of a nephrotoxic agent.
Efavirenz induced liver injury (see section 4.3)
There is some evidence that efavirenz is associated with three clinical pathological patterns of drug induced liver failure in HIV positive patients of which the sub massive necrosis histological pattern seems to be associated with high morbidity/mortality risk and may present many months after therapy has been initiated or even stopped. Risk factors include younger age, CD4+ counts ≥350 cells/µl and female gender. Patients on ATENEF or efavirenz containing antiretroviral treatment (ART) should be regularly monitored for jaundice (including a laboratory bilirubin and liver enzymes) and bleeding tendencies.
Early detection and treatment of the liver failure and the immediate discontinuation of ATENEF or efavirenz containing medicines should be stressed. Patients who discontinue treatment with ATENEF should be followed up for symptoms/signs of liver failure for up to 12 months. ATENEF is not recommended in patients with moderate to severe hepatic impairment because there are insufficient data to determine whether dose adjustments are required. The safety and efficacy of ATENEF in patients with both HIV and hepatitis B virus infection have not been established.
Paediatric Use
ATENEF tablets are not recommended for patients less than 18 years of age because it is a fixed-dose combination tablet containing a component, tenofovir DF, for which safety and efficacy have not been established in this age group.
Use in the elderly
Clinical studies of efavirenz, emtricitabine, or tenofovir DF did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for the elderly patients should be cautious, keeping in mind the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other medicine therapy.
Skin Rash
New-onset skin rash, rash associated with blistering, moist desquamation, or ulceration may occur in patients treated with efavirenz. These include Grade 4 rash (e.g., erythema multiforme, Stevens-Johnson syndrome). Rashes are usually mild-to-moderate maculopapular skin eruptions that occur within the first 2 weeks of initiating therapy with efavirenz and, in most patients continuing therapy with efavirenz, rash resolves within 1 month. ATENEF can be reinitiated in patients interrupting therapy because of rash. ATENEF should be discontinued in patients developing severe rash associated with blistering, desquamation, mucosal involvement, or fever. Appropriate antihistamines and/or corticosteroids may improve the tolerability and hasten the resolution of rash.
Liver Enzymes
In patients with known or suspected history of hepatitis B or C infection and in patients treated with other medications associated with liver toxicity, monitoring of liver enzymes is recommended (see section 4.4, Patients Co-infected With HIV and HBV). In patients with persistent elevations of serum transaminases to greater than five times the ULN, the benefit of continued therapy with ATENEF needs to be weighed against the unknown risks of significant liver toxicity (see section 4.8, Laboratory Abnormalities). Because of the extensive cytochrome P450 mediated metabolism of efavirenz and limited clinical experience in patients with hepatic impairment, caution should be exercised in administering ATENEF to these patients.
Bone Effects
In treatment-naive patients, decreases in bone mineral density (BMD) were seen at the lumbar spine and hip Tenofovir DF is associated with significant increases in biochemical markers of bone metabolism (serum bone-specific alkaline phosphatase, serum osteocalcin, serum C-telopeptide, and urinary N-telopeptide), suggesting increased bone turnover. Serum parathyroid hormone levels and 1,25 Vitamin D levels are also higher in patients receiving tenofovir DF. The effects of tenofovir DF-associated changes in BMD and biochemical markers on long-term bone health and future fracture risk are unknown. For additional information, consult the tenofovir DF package insert.
Cases of osteomalacia (associated with proximal renal tubulopathy) have been reported in association with the use of tenofovir DF (see section 4.8).
Bone monitoring should be considered for HIV infected patients who have a history of pathologic bone fracture or are at risk for osteopenia. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial for all patients. If bone abnormalities are suspected then appropriate consultation should be obtained.
Convulsions
Convulsions have been observed in patients receiving efavirenz, generally in the presence of known medical history of seizures. Caution must be taken in any patient with a history of seizures or psychiatric disorders, including depression.
Patients who are receiving concomitant anticonvulsant medications primarily metabolised by the liver, such as phenytoin and phenobarbital, may require periodic monitoring of plasma levels (see section 4.5).
Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and “cushingoid appearance” have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including the components of ATENEF tablets. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii (carinii) pneumonia (PCP), or tuberculosis), which may necessitate further evaluation and treatment.
Opportunistic infections
Patients receiving ATENEF may continue to develop opportunistic infections and other complications of HIV infection, and therefore they should remain under close observation by medical practitioners experienced in the treatment of patients with associated HIV disease.
The risk of HIV transmission to others
Patients should be advised that current antiretroviral therapy, including ATENEF, has not been proven to prevent the risk of transmission of HIV to others through sexual contact or blood contamination. Appropriate precautions should continue to be employed.
Mitochondrial dysfunction
Nucleoside and nucleotide analogues have been demonstrated in vitro and in vivo to cause a variable degree of mitochondrial damage. There have been reports of mitochondrial dysfunction in HIV negative infants exposed in utero and/or post-natally to nucleoside analogues. The main adverse events reported are haematological disorders (anaemia, neutropenia), metabolic disorders (hyperlactataemia, hyperlipidaemia). These events are often transitory. Some late-onset neurological disorders have been reported (hypertonia, convulsion, abnormal behaviour). Whether the neurological disorders are transient op permanent is not known. Any child exposed in utero to nucleoside and nucleotide analogues, even HIV negative children should have clinical and laboratory follow-up and should be fully investigated for possible mitochondrial dysfunction in case of relevant sign and symptoms.
4.5. Interaction with other medicinal products and other forms of interaction
Interactions (see also section 4.3 and section 4.4)
Efavirenz: Efavirenz has been shown in vivo to induce CYP3A. Other compounds that are substrates of CYP3A may have decreased plasma concentrations when co-administered with efavirenz. In vitro studies have demonstrated that efavirenz inhibits 2C9, 2C19, and 3A4 isozymes in the range of observed efavirenz plasma concentrations. Co-administration of efavirenz with medicines primarily metabolised by these isozymes may result in altered plasma concentrations of the co-administered medicine. Therefore, appropriate dose adjustments may be necessary for these medicines.
Medicines that induce CYP3A4 activity (e.g., phenobarbital, rifampin, rifabutin) would be expected to increase the clearance of efavirenz resulting in lowered plasma concentrations.
Emtricitabine and Tenofovir Disoproxil Fumarate: Since emtricitabine and tenofovir are primarily eliminated by the kidneys, co-administration of ATENEF with medicines that reduce renal function or compete for active tubular secretion may increase serum concentrations of emtricitabine, tenofovir, and/or other renally eliminated medicines. Some examples include, but are not limited to, acyclovir, adefovir dipivoxil, cidofovir, ganciclovir, valacyclovir, and valganciclovir.
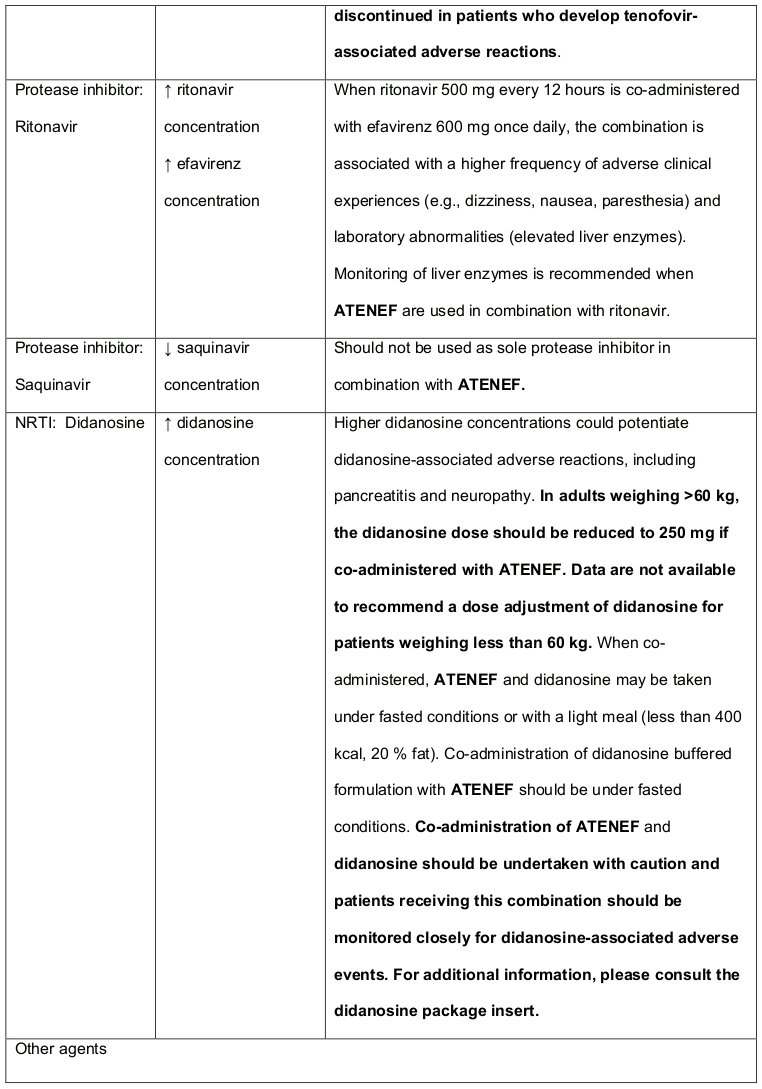
Co-administration of tenofovir DF and didanosine should be undertaken with caution and patients receiving this combination should be monitored closely for didanosine associated adverse events. Didanosine should be discontinued in patients who develop didanosine-associated adverse events (for didanosine dosing adjustment recommendations, see Table 3 in section 4.3). Suppression of CD4+ cell counts has been observed in patients receiving tenofovir DF with didanosine 400 mg daily.
Atazanavir and lopinavir/ritonavir has been shown to increase tenofovir concentrations. The mechanism of this interaction is unknown. Higher tenofovir concentrations could potentiate tenofovir-associated adverse events, including renal disorders. Patients receiving either atazanavir or lopinavir/ritonavir with tenofovir DF (and therefore ATENEF) should be monitored for tenofovir-associated adverse events (for atazanavir dosing adjustment recommendations, see Table 3 in section 4.3).
Other important medicine interaction information for ATENEF is summarised in Table 2 and 3. The medicine interactions described are based on studies conducted with efavirenz, emtricitabine or tenofovir DF as individual agents or are potential medicine interactions; no medicine interaction studies have been conducted using ATENEF. The tables include potentially significant interactions but are not all inclusive.
Table 2. Medicines That Are Contra-indicated or not Recommended for Use With ATENEF:

Table 3. Established and Other Potentially Significant1 Medicine Interactions: Alteration in Dose or Regimen May Be Recommended Based on Medicine Interaction Studies or Predicted Interaction:



Efavirenz Assay Interference
Cannabinoid Test Interaction
Efavirenz does not bind to cannabinoid receptors. False-positive urine cannabinoid test results have been observed in non-HIV-infected volunteers receiving efavirenz when the Microgenics Cedia DAU Multi-Level THC assay was used for screening. Negative results were obtained when more specific confirmatory testing was performed with gas chromatography/mass spectrometry. For more information, please consult the efavirenz package insert.
Other Interactions
Efavirenz
Medicine interaction studies were performed with efavirenz and other medicines likely to be coadministered or medicines commonly used as probes for pharmacokinetic interaction. There was no clinically significant interaction observed between efavirenz and zidovudine, lamivudine, azithromycin, fluconazole, lorazepam, cetirizine, or paroxetine. Single doses of famotidine or an aluminum and magnesium antacid with simethicone had no effects on efavirenz exposures.
Emtricitabine and Tenofovir disoproxil fumarate
No clinically significant medicine interactions have been observed between emtricitabine and famciclovir, indinavir, stavudine, tenofovir disoproxil fumarate and zidovudine. Similarly, no clinically significant medicine interactions have been observed between tenofovir disoproxil fumarate and abacavir, adefovir dipivoxil, efavirenz, emtricitabine, indinavir, lamivudine, lopinavir/ritonavir, methadone, nelfinavir, oral contraceptives, ribavirin and saquinavir/ritonavir in studies conducted in healthy volunteers.
Following multiple dosing to HIV-negative subjects receiving either chronic methadone maintenance therapy, oral contraceptives, or single doses of ribavirin, steady-state tenofovir pharmacokinetics were similar to those observed in previous studies, indicating a lack of clinically significant medicine interactions between these agents and tenofovir disoproxil fumarate.
Immunosuppressants metabolised by CYP3A4 (e.g. ciclosporin, sirolimus)/Efavirenz
Dose adjustments of the immunosuppressant may be required. Close monitoring of immunosuppressant concentrations for at least two weeks (until stable concentrations are reached) is recommended when starting or stopping treatment with ATENEF.
4.6. Fertility, pregnancy and lactation
Pregnancy
ATENEF should not be used in pregnancy.
Efavirenz may cause foetal harm when administered during the first trimester to a pregnant woman. Pregnancy should be avoided in women receiving ATENEF. Barrier contraception must always be used in combination with other methods of contraception (e.g., oral or other hormonal contraceptives). Women of childbearing potential should undergo pregnancy testing before initiation of ATENEF. If this medicine is used during the first trimester of pregnancy, or if the patient becomes pregnant while taking this medicine, the patient should be appraised of the potential harm to the foetus. Birth defects may occur in live births.
There are no adequate and well-controlled studies of ATENEF.
Breastfeeding
It is recommended that HIV-1 infected mothers not breast-feed their infants to avoid risking postnatal transmission of HIV. Studies in rats have demonstrated that both efavirenz and tenofovir are secreted in milk. It is not known whether efavirenz, emtricitabine, or tenofovir is excreted in human milk. Because of both the potential for HIV transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breast-feed if they are receiving ATENEF.
Fertility
No human data on the effect of ATENEF are available.
4.7. Effects on ability to drive and use machines
Patients who experience central nervous system symptoms such as dizziness, impaired concentration, and/or drowsiness should avoid potentially hazardous tasks such as driving or operating machinery.
4.8. Undesirable effects
The undesirable effects from clinical study and post-marketing experience with ATENEF and the individual components of ATENEF in antiretroviral combination therapy are listed in the table below by body system organ class, frequency and the component(s) of ATENEF to which the undesirable effects are attributable.
Tabulated list of adverse reactions:
| ATENEF | |||
|---|---|---|---|
| Efavirenz | Emtricitabine | Tenofovir disoproxil fumarate | |
| Blood and lymphatic system disorders | |||
| Frequent | neutropenia | ||
| Less frequent | anaemia | neutropenia | |
| Immune system disorders | |||
| Frequent | allergic reaction | allergic reaction | |
| Less frequent | hypersensitivity Immuno- allergic liver injury/failure | angioedema | angioedema |
| Metabolism and nutrition disorders | |||
| Frequent | hyperglycaemia, hypertriglycerid-aemia lactic acidosis, usually associated with severe hepatomegaly and steatosis | hypophosphataemia, hyperglycaemia, hypertriglyceridaemia | |
| Less frequent | raised serum cholesterol and triglycerides | hypokalaemia lactic acidosis, usually associated with severe hepatomegaly and steatosis | |
| Psychiatric disorders | |||
| Frequent | depression, anxiety, abnormal dreams,insomnia | abnormal dreams, insomnia, depressive disorders | depression |
| Less frequent | suicide attempt, suicide ideation, psychosis, mania, paranoia, hallucination, euphoric mood, affect lability, confusional state, aggression completed suicide, delusion, neurosis, depersonalisation | ||
| Nervous system disorders | |||
| Frequent | cerebellar coordination and balance disturbances, somnolence, headache, disturbance in attention, dizziness | headache dizziness | dizziness headache, insomnia, peripheral neuropathy including peripheral neuritis, anxiety |
| Less frequent | convulsions, amnesia, thinking abnormal, ataxia, coordination abnormal, agitation, tremor, paraesthesia, hypoaesthesia | asthenia, sleep disorders, neuropathy, peripheral neuritis, paraesthesia | |
| Eye disorders | |||
| Less frequent | vision blurred | ||
| Ear and labyrinth disorders | |||
| Less frequent | tinnitus, vertigo | ||
| Cardiac disorders | |||
| Less frequent | palpitations | ||
| Vascular disorders | |||
| Less frequent | flushing | ||
| Respiratory, thoracic and mediastinal disorders | |||
| Frequent | dyspnoea | increased cough, rhinitis | chest pain, pneumonia, dyspnoea |
| Gastrointestinal disorders | |||
| Frequent | diarrhoea, vomiting, abdominal pain, nausea anorexia, constipation, malabsorption, dyspepsia | diarrhoea, nausea elevated amylase including elevated pancreatic amylase, elevated serum lipase, vomiting, abdominal pain, dyspepsia | diarrhoea, vomiting, nausea abdominal pain, abdominal distension, flatulence, raised serum amylase, anorexia, pancreatitis |
| Less frequent | pancreatitis, raised serum amylase | ||
| Hepatobiliary disorders | |||
| Frequent | Raised liver enzymes | elevated serum aspartate aminotransferase (AST) and/or elevated serum alanine aminotransferase (ALT), hyperbilirubinaemia | increased transaminases |
| Less frequent | hepatitis acute hepatic failure | hepatic steatosis, hepatitis | |
| Skin and subcutaneous tissue disorders | |||
| Frequent | rash (moderate-severe, all grades), pruritus | vesiculobullous rash, pustular rash, maculopapular rash, rash, pruritus, urticaria, skin discolouration (increased pigmentation) | rash (including pruritus, maculopapular rash, urticaria,vesiculo-bullous rash and pustular rash) |
| Less frequent | Stevens-Johnson syndrome, erythema multiforme, severe rash, photoallergic dermatitis | ||
| Musculoskeletal and connective tissue disorders | |||
| Frequent | arthralgia, myalgia | elevated creatine kinase | |
| Less frequent | myopathy | arthralgia, myalgia | rhabdomyolysis, muscular weakness osteomalacia (manifested as bone pain and infrequently contributing to fractures), myopathy, myalgia, arthralgia |
| Renal and urinary disorders | |||
| Less frequent | increased creatinine, proteinuria renal failure (acute and chronic), acute tubular necrosis, proximal renal tubulopathy including Fanconi syndrome, nephritis (including acute interstitial nephritis), nephrogenic diabetes insipidus | ||
| Reproductive system and breast disorders | |||
| Less frequent | gynaecomastia | ||
| General disorders and administration site conditions | |||
| Frequent | fatigue, asthenia | pain, asthenia | asthenia |
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows continued monitoring of the benefit/risk balance of the medicine. Health care providers are asked to report any suspected adverse reactions to SAHPRA via the “6.04 Adverse Drug Reactions Reporting Form”, found online under SAHPRA’s publications: https://www.sahpra.org.za/Publications/Index/8.
6.2. Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.