SIGNIFOR LAR Suspension for injection Ref.[27986] Active ingredients: Pasireotide
Source: Health Products and Food Branch (CA) Revision Year: 2020
Action and clinical pharmacology
Mechanism of Action
Pasireotide is a second generation cyclohexapeptide, injectable somatostatin analogue. Like the natural peptide hormones somatostatin-14 and somatostatin-28 [also known as somatotropin release inhibiting factor (SRIF)], pasireotide exerts its pharmacological activity via binding to somatostatin receptors (SSTRs). There are five known human somatostatin receptor subtypes: SSTR 1, 2, 3, 4, and 5. These receptor subtypes are expressed in different tissues under normal physiological conditions. Somatostatin analogues bind to SSTR receptors with different potencies (Table 5). Pasireotide binds with high affinity to four of the five SSTRs: SSTR5˃SSTR2˃SSTR3˃SSTR1.
Table 5. Binding Affinities of Somatostatin (SRIF-14) and Pasireotide to the Five Human SSTR Receptor Subtypes (SSTR1-5):
| Compound | SSTR1 | SSTR2 | SSTR3 | SSTR4 | SSTR5 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Somatostatin (SRIF-14) | 0.93±0.12 | 0.15±0.02 | 0.56±0.17 | 1.5±0.4 | 0.29±0.04 | Pasireotide | 9.3±0.1 | 1.0±0.1 | 1.5±0.3 | >100 | 0.16±0.01 |
Results are the mean +SEM of IC50 values expressed as nmol/L (nM).
Pharmacodynamics
Somatostatin receptors are expressed in many tissues, especially in neuroendocrine tumors (e.g., growth hormone (GH) or adrenocorticotrophic hormone (ACTH) secreting pituitary adenomas), in which hormones are excessively secreted (e.g., GH in acromegaly and ACTH in Cushing's disease).
Acromegaly
Pasireotide binds to SSTR2 and SSTR5 subtype receptors which may be relevant for inhibition of GH secretion.
Cushing's disease
In vitro studies have shown that corticotroph tumor cells from Cushing's disease patients display a high expression of SSTR5, whereas the other receptor subtypes are either not expressed or are expressed at lower levels. Pasireotide binds and activates the SSTRs of the corticotrophs in ACTH producing adenomas resulting in inhibition of ACTH secretion. The high affinity of pasireotide for four of the five SSTRs, especially to SSTR5, provides the basis for pasireotide to be an effective treatment for Cushing's disease patients.
Cardiac Electrophysiology
The effects of pasireotide (administered subcutaneously; referred to as SIGNIFOR s.c.) on cardiac electrophysiology were assessed in two dedicated ECG assessment studies (see WARNINGS AND PRECAUTIONS – Cardiovascular and Monitoring and Laboratory Tests, ADVERSE REACTIONS, and DRUG INTERACTIONS).
ECG Study 1: In the first randomised, double-blind, placebo-controlled, crossover ECG assessment study, healthy volunteers (N=77) received treatment for 4 days with a supratherapeutic subcutaneous dose of pasireotide 1950 μg BID, followed by a 1950 μg morning dose on day 5. ECG assessments were performed at 10 time points on day 5. SIGNIFOR 1950 μg treatment was associated with statistically significant decreases in heart rate and prolongation of the Fridericia-corrected QT interval (QTcF=QT/RR0.33) at all timepoints on day 5. The maximum placebo-adjusted mean changes from baseline occurred at 2 h post-dosing and were -12.6 bpm (90% CI -13.9, -11.3) for heart rate and 17.5 ms (90% CI 15.5, 19.4) for the QTcF interval. SIGNIFOR 1950 μg treatment was also associated with statistically significant increases in the PR interval, with a maximum placebo-adjusted mean change from baseline of 6.9 ms (90% CI 5.4, 8.5) at 4 h post-dosing.
ECG Study 2: In a second randomised, double-blind, placebo-controlled, crossover ECG assessment study in healthy volunteers (N=105), subjects received treatment for 4 days with a therapeutic s.c. dose of SIGNIFOR 600 μg BID and a supratherapeutic subcutaneous dose of pasireotide 1950 μg BID, followed by 600 μg and 1950 μg morning doses on day 5. ECG assessments were performed at 11 time points on day 5.
In both the 600 μg and 1950 μg treatment arms, pasireotide was associated with statistically significant QTcF prolongation at all timepoints on day 5. The maximum placebo-adjusted mean change from baseline occurred at 2 h post-dosing in both treatment arms and was 11.8 ms (90% CI 10.0, 13.5) in the 600 μg treatment arm and 14.0 ms (90% CI 12.3, 15.8) in the 1950 μg arm.
The mechanism for the observed QT prolongation is not known.
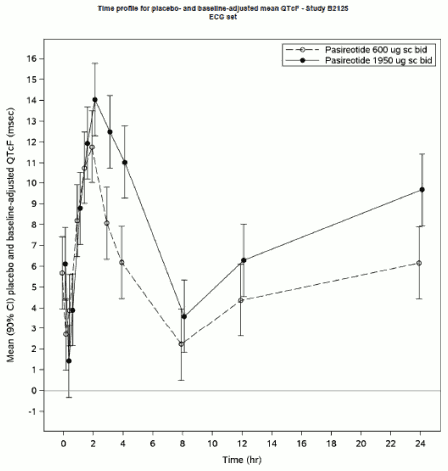
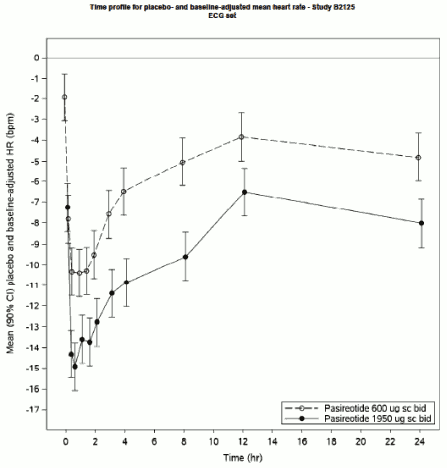
In both the 600 μg and 1950 μg treatment arms, SIGNIFOR s.c. was associated with statistically significant reductions in heart rate at all timepoints on day 5. The maximum placebo-adjusted mean change from baseline was -10.4 bpm (90% CI -11.5, -9.2) at 1 h post-dosing in the 600 μg treatment arm and -14.9 bpm (90% CI -16.1, -13.8) in the 1950 μg arm.
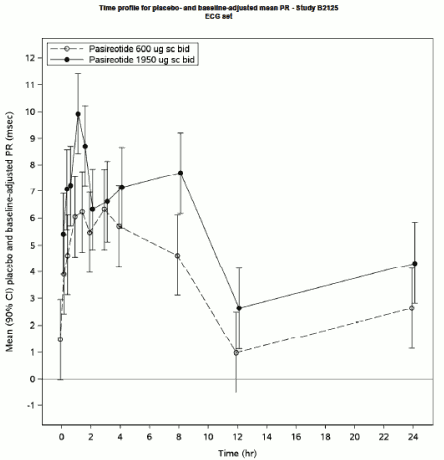
Statistically significant prolongation of the PR interval occurred from 0.25 to 8 h post-dosing in the SIGNIFOR s.c. 600 μg arm and at all timepoints in the SIGNIFOR s.c. 1950 μg arm on day 5. The maximum placebo-adjusted mean change from baseline occurred at 2 h post-dosing in both treatment arms and was 6.1 ms (90% CI 4.6, 7.6) in the 600 μg arm and 9.9 ms (90% CI 8.4, 11.4) in the 1950 μg arm.
On the basis of population pharmacokinetic modelling, the predicted steady-state mean peak plasma concentration for the maximum SIGNIFOR LAR dose of 60 mg dose every 28 days in acromegaly patients would be 25.8 ng/mL, which is similar to the observed steady-state mean peak concentration (24.3 mg/mL) of SIGNIFOR s.c. 600 μg twice a day and below the observed steady-state mean peak concentration (80.6 ng/mL) of the 1,950 μg twice a day dose in ECG Study 2. The predicted peak concentrations for the maximum SIGNIFOR LAR dose of 40 mg in Cushing's disease patients is 14 ng/mL, which is below the observed peak concentrations of Signifor s.c. described above.
Pharmacokinetics
Table 6. Summary of SIGNIFOR LAR (monthly dosing) Pharmacokinetic Parameters in Acromegaly Patients:
| Dose (mg) | Ctrough,ss (ng/mL)* | Cmaxp2,ss (ng/mL)* | Tmax,p2 (days)** | AUC0-28d,ss (hr*ng/mL)* |
|---|---|---|---|---|
| 20 | 3.77 (1.94) | 5.04 (2.00) | 21 | 2749 (1099) |
| 40 | 7.16 (3.13) | 8.03 (3.17) | 21 | 4788 (1974) |
| 60 | 13.3 (8.9) | 17.8 (8.9) | 22 | 8700 (3822) |
* Data expressed as mean (SD) values and from the third dose at steady state;
** Data expressed as median values from the first dose.
Pasireotide for intramuscular use is formulated as microspheres for long-acting release. After a single injection, the plasma pasireotide concentration shows an initial burst release on the injection day, followed by a dip from Day 2 to Day 7, then a slow increase to the maximum concentration around Day 21, and a slow declining phase over the next weeks, concomitant with the terminal degradation phase of the polymer matrix of the dosage form.
Absorption
No studies have been conducted to evaluate the absolute bioavailability of pasireotide in humans. Food effect is unlikely to occur since SIGNIFOR LAR is administered via the parenteral route. The relative bioavailability of SIGNIFOR LAR compared to SIGNIFOR s.c. is 106-148%.
Distribution
In healthy volunteers, pasireotide administered as SIGNIFOR LAR is widely distributed with a large apparent volume of distribution (Vz/F >100 L). Distribution between blood and plasma is concentration independent and shows that pasireotide is primarily located in the plasma (91%). Plasma protein binding is moderate (88%) and independent of concentration.
Metabolism
Pasireotide was shown to be highly metabolically stable. In healthy volunteers, pasireotide in its unchanged form is the predominant form found in plasma, urine, and feces.
Excretion
Pasireotide is eliminated mainly via hepatic clearance (biliary excretion) with a small contribution of the renal route. In a human ADME study with pasireotide administered as SIGNIFOR s.c. with a single dose of 600 microgram, 55.9 ± 6.63% of the radioactivity dose was recovered over the first 10 days post dosing, including 48.3 ± 8.16% of the radioactivity in feces and 7.63 ± 2.03% in urine.
The apparent clearance (CL/F) of pasireotide administered as SIGNIFOR LAR in healthy volunteers is on average 4.5 to 8.5 L/h. Based on population pharmacokinetic (PK) analyses, the estimated CL/F was approximately 4.8 to 6.5 L/h for typical Cushing's disease patients, and approximately 5.6 to 8.2 L/h for typical acromegaly patients.
Steady-state pharmacokinetics, Dose proportionality, and Accumulation
Following monthly (every 28 days) i.m. injections of 20 mg, 40 mg, and 60 mg SIGNIFOR LAR, a steady state was achieved after three monthly doses in acromegaly patients. PK exposures of SIGNIFOR LAR were approximately dose proportional (steady-state trough concentration; Ctrough,ss) from 10 mg to 60 mg in patients. Peak-to-trough ratio (<1.5) and accumulation (ratio of 1.5-2.0) of SIGNIFOR LAR were low to moderate on multiple dosing.
Special Populations and Conditions
Pediatrics (<18 years of age): No studies have been performed in pediatric patients (see INDICATIONS AND CLINICAL USE, WARNINGS AND PRECAUTIONS – Special Populations, Pediatrics, and DOSAGE AND ADMINISTRATION - Special Populations, Pediatrics).
Geriatrics (≥65 years of age): Age is not a significant covariate in the population PK analysis of patients. Therefore age is not expected to significantly impact circulating levels of pasireotide. Efficacy and safety data on patients older than 65 years are limited (see INDICATIONS AND CLINICAL USE, WARNINGS AND PRECAUTIONS – Special Populations, Geriatrics, and DOSAGE AND ADMINISTRATION - Special Populations, Geriatrics).
Gender: Female acromegaly patients had a higher exposure of 32% and 51% compared to male patients in studies with medical treatment naïve patients and inadequately controlled patients, respectively; these differences in exposure were not clinically relevant based on efficacy and safety data. Population PK analyses of pasireotide administered as SIGNIFOR LAR in Cushing's disease patients demonstrates that gender is not a significant covariate and suggests that gender does not influence PK parameters. No dose adjustment is required based on gender.
Race: Population PK analyses of pasireotide administered as SIGNIFOR LAR suggest that race does not have clinically relevant influence on PK parameters. No dose adjustment is required based on race.
Hepatic Insufficiency: SIGNIFOR LAR is contraindicated in patients with moderate or severe hepatic insufficiency (see CONTRAINDICATIONS). In a clinical study with single dose of pasireotide administered as SIGNIFOR s.c. in subjects with impaired hepatic function (ChildPugh A, B and C), subjects with moderate and severe hepatic impairment (Child-Pugh B and C) showed significantly higher exposures than subjects with normal hepatic function. Upon correction for covariate effect (age, BMI, and albumin) AUCinf was increased by 60% and 79%, Cmax increased by 67% and 69%, and CL/F decreased by 37% and 44%, respectively, in the moderate and severe hepatic impairment groups relative to the control group.
Renal Impairment: No dose adjustment is required in patients with mild or moderate impaired renal function. SIGNIFOR LAR should be used with caution in patients with severe renal impairment.
In a clinical study with single dose administration of 900 µg pasireotide as SIGNIFOR s.c. in subjects with impaired renal function, the degree of renal impairment did not have a significant impact on the pharmacokinetics of pasireotide. The AUC0-inf decreased by 22%, 14%, and 1% for mild, moderate, and severe renally impaired subjects and increased by 25% in ESRD subjects compared to normal subjects adjusted for age, gender, and weight as covariates. The Cmax decreased by 28%, 23%, 19%, and 10% for mild, moderate, severe renally impaired, and ESRD subjects compared to normal subjects adjusted for age, gender, and weight as covariates.
However, increases in unbound pasireotide AUCinf,u of 1.85, 2.41, 2.96 fold and Cmax,u of 1.36, 2.00, 3.01 fold were observed in patients with moderate, severe renal impairment, and ESRD. Grade 3 and Grade 4 increases in amylase, lipase, and uric acid, and grade 3 decreases in hemoglobin were also observed in subjects with severe renal impairment and ESRD. Hence, caution is recommended for the use of pasireotide in patients with severe renal impairment and ESRD (see WARNINGS AND PRECAUTIONS – Renal, WARNINGS AND PRECAUTIONS - Monitoring and laboratory tests, and DOSAGE AND ADMINISTRATION, Renal impairment).
Body weight: PK exposures had a slight correlation with body weight in the study with medical treatment naïve patients (approximately 39% increase for 40 kg decrease in body weight), but not in the study with inadequately controlled patients. These changes are not clinically significant. No dose adjustment is required based on body weight.
Genetic polymorphism: The effect of genetic polymorphism on the pharmacokinetics of SIGNIFOR LAR has not been established.
Detailed pharmacology
Nonclinical Pharmacology
In vitro pharmacology
Pasireotide is a somatostatin analogue with high binding affinity and high functional activity for somatostatin receptor subtypes sst1, 2, 3 and sst5. In contrast, octreotide primarily bound with high sub-nanomolar affinity to the sst2 subtype and with lower affinities to the sst3 and sst5 subtype. In rat primary GH secreting pituitary cells in vitro, pasireotide was 3 times more potent than octreotide to reduce growth hormone releasing hormone (GHRH)-induced GH secretion.
In vivo pharmacology
In several experiments investigating the short term and long term effects of pasireotide in vivo after s.c. injection or after application via osmotic minipumps or by using the long acting release formulation (LAR) a strong and long lasting effect on plasma levels of GH and IGF-1 was found.
Nonclinical Pharmacokinetics
Pasireotide is well absorbed after s.c. dosing in all species tested with complete bioavailability. Bioavailability for LAR was estimated also to be ~100% in rats after i.m. dosing. Plasma exposures were generally proportional to the dose in rats. The plasma protein binding is moderate across species with the lowest binding in human (88%); therefore a substantial change in drug kinetics due to protein binding changes is not expected. Pasireotide and/or its metabolites in tissues were eliminated slowly and were mainly distributed to the adrenal cortex, kidney cortex, bone marrow, blood vessel wall, lymph nodes, spleen, and liver while showing minimal brain penetration and no specific retention in melanin-rich tissues (uveal tract and skin). Pasireotide and/or its metabolites showed some distribution to the fetus in rats and rabbits. The transfer of pasireotide-related radioactivity into milk was observed in rats.
Human Pharmacology
Pharmacodynamics
Clinical studies in patients with acromegaly have shown that GH and IGF-1 levels markedly decrease by Month 3 with monthly intramuscular (i.m.) injections of SIGNIFOR LAR varying from 20 mg to 60 mg and the reduction is sustained overtime.
PK/PD analyses for efficacy based on Emax model and logistic regression model have shown that higher exposure of pasireotide is associated with greater suppression of GH and IGF-1 levels, and that pasireotide is superior to octreotide in terms of suppression of IGF-1. The results of the PK/PD analyses are consistent with the GH and IGF-1 response rates observed in studies C2305 and C2402.
PK/PD analyses for safety have shown that the odds of post-baseline hyperglycemia increases moderately with increasing pasireotide exposure from 40 mg to 60 mg. In addition, higher baseline HbA1c and higher baseline FPG are associated with a higher probability of developing hyperglycemia. No correlation was found between pasireotide exposures and QT or liver functions.
Pharmacokinetics
After a single intramuscular (i.m.) injection of SIGNIFOR LAR varying from 10 mg to 60 mg in healthy volunteers, PK profiles showed an initial burst release (Cmax,p1) at 12 hours post injection on Day 1 (Tmax,p1), followed by a dip from Day 2 to Day 7, then a slow increase to maximum concentration (Cmax,p2) at Day 20 (Tmax,p2), followed by a declining phase over the next seven weeks. The Cmax,p2 and AUCinf for 60 mg SIGNIFOR LAR dose, range between 15.8-29 ng/mL and 7971-13395 ng.hr/mL, respectively. The PK profiles of SIGNIFOR LAR were similar between Western and Asian subjects. PK exposures (Cmax,p2 and AUCinf) were approximately dose proportional from 10 mg to 60 mg. The apparent clearance (CL/F) of pasireotide LAR was 4.5 to 8.5 L/hr. The apparent volume of distribution (Vz/F) was large (>100 L). The apparent terminal half-life (T1/2) for SIGNIFOR LAR was approximately 16 days. The relative bioavailability of SIGNIFOR LAR to SIGNIFOR was 106-148%. Pharmacokinetics (PK) is comparable between healthy volunteers and acromegaly patients. PK exposures reached steady state after 3 injections (see ACTION AND CLINICAL PHARMACOLOGY).
Toxicology
Safety pharmacology
The cardiovascular safety of pasireotide was evaluated using in vitro methods (hERG assay, Purkinje fiber assay, effect of pasireotide on potassium channel currents, effect of pasireotide on sodium and calcium channel currents) and in an in vivo conscious monkey telemetry study (single s.c. doses of 0.4, 0.8 and 1.6 mg/kg). In vitro, a statistically significant, concentrationdependent hERG current block was observed at 13 and 39 µg/mL (8.2% and 16.8% inhibition, respectively). In vivo, a NOAEL of 1.6 mg/kg was obtained in monkeys (N=4).
The effect of pasireotide on respiratory function and neurobehaviour was assessed in rats (single s.c. doses of 0.8, 1.6 and 4.0 mg/kg) and mice (single s.c. doses of 0.4, 1.2, 4.0 and 12 mg/kg), respectively. Toxicologically significant effects were not observed.
Single dose toxicity
The acute toxicity of pasireotide was assessed in rats and mice at 15 and 30 mg/kg by the s.c. route. Lethalities were not observed.
Repeated dose toxicity
Rats
The pivotal rodent repeat-dose toxicity study was conducted in male and female rats. Animals were administered pasireotide by s.c. injection once daily at 0.0008, 0.024, 0.08 and 0.24 mg/kg/day for 26 weeks. When compared with human AUC values at 900 µg bid, these dose levels provide an exposure margin of 0.07, 0.24 (0.33 and 0.15 for males and females, respectively), 0.49, 1.92, respectively. The NOAEL was considered to be 0.024 mg/kg/day based on histological alterations in the pituitary (males) and the genital tract (females). Additionally, in the 26 week (6 cycle) rat study, i.m. administration of 3.125 and 6.25 mg pasireotide LAR led to AUC0-28d of 620.8 and 1062.5 ng.d/mL after the first cycle, leading to exposure multiples of 1.3 and 2.3-fold for AUC0-28d over the systemic exposure at human dose of 60 mg.
All pasireotide-mediated effects were considered a result of the drug's pharmacology and all changes demonstrated reversibility following a drug-free period. Decreased body weight was observed in males (from 0.008 mg/kg) and females (0.24 mg/kg). In males, decreased pituitary weight and decreased cytoplasmic mass of acidophile cells/somatotrophs was observed at doses >0.024 mg/kg. In females, alterations in the genital tract (decreased number of corpora lutea, vaginal mucosal hyperplasia or hypertrophy of mucification, vaginal hypertrophy) consistent with prolongation of the estrous cycle were observed at doses ≥0.08 mg/kg.
Inhibitory effects on lymphoid and hematopoietic organs were observed and included decrease thymus weight and cellularity as well as decreased hematopoietic activity of the spleen and bone marrow. A lack of new bone formation beneath the epiphyseal plate of the tibia and femur was observed. Serum biochemistry changes (increased ALT, decreased albumin) and decreased liver weight suggested possible effects on the liver at high dose levels, possibly as a secondary result to the decrease in IGF-1. Changes in coagulation parameters (increased PT and APTT) noted in females are likely related to the pharmacologic effect of pasireotide, probably through modification of the liver production of coagulating factors regulated by GH.
The results of the pasireotide LAR studies demonstrated systemic effects that are similar to that by the s.c. formulation. The microscopic changes in target organs (pituitary, adrenal and thyroid glands, pancreas, bones and bone marrow) are consistent with the pharmacologic activity of somatostatin analogues.
Monkeys
The pivotal non-rodent repeat-dose toxicity study was conducted in male and female monkeys. Animals were administered pasireotide at 0.4, 1.6, and 3.2 mg/kg/day for 39 weeks. When compared with human AUC values at 900 µg bid, these dose levels provide an exposure margin of 12.2, 39.0 and 96.1 for males, and 13.3, 54.7 and 102.6 for females. The NOAEL was considered to be 1.6 mg/kg/day based on histological alterations in the pituitary (increased acidophilia in the pars distallis), thyroid (small follicles), large intestine (distension with firm fecal material), and injection site reactions. All pasireotide-mediated effects were considered a result of the drug's pharmacology and all changes demonstrated reversibility following a drugfree period.
Genotoxicity
Pasireotide did not exhibit mutagenic or clastogenic potential in a battery of assays including the Ames test, human peripheral lymphocyte chromosome aberration test, or the in vivo rat micronucleus test.
Carcinogenicity
The carcinogenic potential of pasireotide was assessed by the s.c. route in the 26-week transgenic RasH2 mouse model (dose levels: 0, 0.5, 1.0, 2.5 mg/kg/day) and the 2-year rat bioassay (dose levels: 0, 0.01, 0.05, 0.3 mg/kg/day). Pasireotide was not carcinogenic in either model.
Reproductive and Developmental Toxicity
Fertility and early embryonic development were evaluated in rats. Pasireotide was administered by s.c. injection at 0.1, 1.0 and 10 mg/kg/day prior to mating, during mating, and through gestation day (GD) 6. Reproductive effects were observed in females only and included prolonged estrus cycles/acyclicity at doses ≥1.0 mg/kg and decreased numbers of corpora lutea, implantation sites, and/or viable fetuses at all doses. A NOAEL for female fertility was not established (<0.1 mg/kg/day).
Embryo-fetal development was evaluated in rats and rabbits. In rats, pasireotide was administered by s.c. injection at 1, 5, and 10 mg/kg/day from GD 6-17. At 10 mg/kg, and in the presence of maternal toxicity and mortality, effects on the F1 generation were noted and consisted of increased early/total resorptions, decreased fetal weights, and mal-rotated limbs. The fetal NOAEL was 5 mg/kg. Pasireotide was not teratogenic in the rat.
In rabbits, pasireotide was administered by s.c. injection at 0.05, 1.0 and 5.0 mg/kg/day from GD 7-20. Maternal toxicity was observed from 1.0 mg/kg and mortality occurred at 5.0 mg/kg. Reproductive and fetal effects (increased early and/or total resorptions, decreased fetal weights) were noted in the presence of maternal toxicity at doses ≥1 mg/kg. At 5 mg/kg, abortions and a decreased number of viable fetuses were seen. Increased skeletal variations noted at 5.0 mg/kg were considered secondary to the reduced fetal weights. The maternal and fetal NOAEL were 0.05 mg/kg. Pasireotide was not teratogenic in the rabbit.
Pre- and post-natal development were evaluated in rats. Pasireotide was administered by s.c. injection at 2, 5, and 10 mg/kg/day to F0 generation dams from GD 6 to day 21, 22, or 23 post partum. Maternal toxicity was observed at all doses and drug-related mortality was noted at 5 mg/kg. Maternal performance was unaffected by administration of pasireotide (no change in gestation index, length of gestation, numbers of live, dead pups, number of implantation scars, sex ratio and the live birth index). Lower F1 body weights were seen at all doses. Secondary to the lower pup weights, the mean day of pinna unfolding was slightly increased in all dose groups. Post weaning, body weight gains were comparable for all groups demonstrating reversibility. There was no effect on visual function, physical development, behavioural performance, macroscopic findings, parental performance or uterine findings for the F1 generation adults.
Antigenicity
Antigenicity was not evaluated with the s.c. formulation. Using pasireotide LAR in a rat i.m. study, anti-pasireotide antibodies were detected in 26/59 treated animals. The antibodies were considered non-neutralizing, as pharmacologic effects and drug levels were sustained.
Immunotoxicity
The immunotoxic potential of pasireotide was evaluated in a 4-week rat s.c. immunotoxicity study (dose levels: 0.08, 0.24, and 0.8 mg/kg/day). Pasireotide exhibits low immunotoxic potential. Although a slight decrease in lymphocytes counts was observed in males at 0.24 and 0.8 mg/kg/day (total lymphocyte counts and absolute counts of Total T lymphocytes, Helper T lymphocytes, Cytotoxic T lymphocytes, natural killer lymphocytes and B lymphocytes), there were no toxicologically-relevant pasireotide effects on immune function (anti-KLH IgM, anti-KLH IgG responses unaffected by pasireotide treatment).
Phototoxicity
In the absorption spectrum of pasireotide, a significant peak was found at around 360 nm. An in vitro phototoxicity assay was performed. Pasireotide did not exhibit phototoxic potential.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.