XEOMIN Powder for solution for injection Ref.[11109] Active ingredients: Botulinum toxin type A
Source: FDA, National Drug Code (US) Revision Year: 2021
12.1. Mechanism of Action
XEOMIN blocks cholinergic transmission at the neuromuscular and salivary neuroglandular junction by inhibiting the release of acetylcholine from peripheral cholinergic nerve endings. This inhibition occurs according to the following sequence: neurotoxin binding to cholinergic nerve terminals, internalization of the neurotoxin into the nerve terminal, translocation of the light-chain part of the molecule into the cytosol of the nerve terminal, and enzymatic cleavage of SNAP25, a presynaptic target protein essential for the release of acetylcholine. In both muscles and glands, impulse transmission is re-established by the formation of new nerve endings.
12.3. Pharmacokinetics
Using currently available analytical technology, it is not possible to detect XEOMIN in the peripheral blood following intramuscular or intraglandular injection at the recommended doses.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Studies to evaluate the carcinogenic potential of XEOMIN have not been conducted.
Mutagenesis
Genotoxicity studies have not been conducted for XEOMIN.
Impairment of Fertility
In a fertility and early embryonic development study in rabbits, males and females were dosed with XEOMIN (1.25 Units/kg, 2.5 Units/kg, or 3.5 Units/kg) intramuscularly every two weeks for 5 and 3 doses, respectively, beginning 2 weeks prior to mating. No effects on mating or fertility were observed. The highest dose tested is approximately twice the maximum recommended human dose for cervical dystonia (120 Units) on a body weight basis.
14. Clinical Studies
14.1 Chronic Sialorrhea
Chronic Sialorrhea in Adult Patients
The efficacy and safety of XEOMIN for the treatment of chronic sialorrhea in adult patients were evaluated in a double-blind, placebo-controlled clinical trial that enrolled a total of 184 patients with chronic sialorrhea resulting from Parkinson's disease, atypical parkinsonism, stroke, or traumatic brain injury, that was present for at least three months. Patients with a history of aspiration pneumonia, amyotrophic lateral sclerosis, salivary gland or duct malformation, and gastroesophageal reflux disease were excluded. The study consisted of a 16-week main phase, followed by an extension period of dose-blinded treatment with XEOMIN.
In the main phase, a fixed total dose of XEOMIN (100 Units or 75 Units) or placebo was administered into the parotid and submandibular salivary glands in a 3:2 dose ratio. The co-primary efficacy variables were the change in unstimulated Salivary Flow Rate (uSFR, Table 14) and the change in Global Impression of Change Scale (GICS, Table 15) at Week 4 post-injection. A total of 173 treated patients completed the main phase of the study. For both the uSFR and GICS, XEOMIN 100 Units was significantly better than placebo (see Table 14 and Table 15). XEOMIN 75 Units was not significantly better than placebo.
Table 14. Mean Change in uSFR (g/min) from Baseline at Week 4, 8, 12, and 16 of Main Phase:
| XEOMIN 100 Units | Placebo | |
|---|---|---|
| N=73 | N=36 | |
| Week 4* | -0.13 | -0.04 |
| Week 8 | -0.13 | -0.02 |
| Week 12 | -0.12 | -0.03 |
| Week 16 | -0.11 | -0.01 |
* p=0.004
Table 15. Mean GICS at Week 4, 8, 12, and 16 of Main Phase:
| XEOMIN 100 Units | Placebo | |
|---|---|---|
| N=74 | N=36 | |
| Week 4* | 1.25 | 0.67 |
| Week 8 | 1.30 | 0.47 |
| Week 12 | 1.21 | 0.56 |
| Week 16 | 0.93 | 0.41 |
* p=0.002
In the extension period, patients received up to three additional treatments with XEOMIN 100 Units or 75 Units every 16±2 weeks, for a total exposure duration of up to 64 weeks. Patients had periodic dental examinations to monitor for changes in dentition and oral mucosa. A total of 151 patients completed the extension period.
Chronic Sialorrhea in Pediatric Patients
The efficacy and safety of XEOMIN for the treatment of chronic sialorrhea in pediatric patients were evaluated in a prospective, randomized, double-blind, placebo-controlled (ages 6-17 years), parallel-group, multicenter trial that enrolled and treated a total of 216 pediatric patients 6-17 years of age with chronic sialorrhea associated with cerebral palsy, other genetic or congenital disorders, or traumatic brain injury. An additional 35 patients 2-5 years of age were treated with open-label XEOMIN in that study. The study consisted of a 16-week main phase, followed by an open-label extension period of treatment with XEOMIN where patients could receive up to 3 additional treatments with XEOMIN every 16 ± 2 weeks, for a total exposure duration of up to 64 weeks (222 patients completed the extension period).
In the main phase, patients 6-17 years of age were administered a total dose of XEOMIN according to body weight (up to 75 Units), or placebo, into the parotid and submandibular glands in a 3:2 dose ratio, using ultrasound guidance. Patients 2-5 years of age all received open-label treatment with XEOMIN, according to body weight, using ultrasound guidance. Patients with a body weight <12 kg were excluded.
The primary efficacy analysis was conducted in the 6-17 years of age patient group. The co-primary endpoints were the change in unstimulated Salivary Flow Rate (uSFR, Table 16) and carer's Global Impression of Change Scale (GICS, Table 17) at Week 4 post-injection.
For both the uSFR and GICS, XEOMIN was statistically significantly better than placebo (see Table 16 and Table 17).
Table 16. Mean change in uSFR (g/min) from Baseline at Week 4, 8, 12, and 16 of Main Phase:
| XEOMIN (6-17 years) N=148 | Placebo (6-17 years) N=72 | |
|---|---|---|
| Week 4* | -0.14 | -0.07 |
| Week 8 | -0.16 | -0.07 |
| Week 12 | -0.16 | -0.06 |
| Week 16 | -0.15 | -0.08 |
* p=0.0012
Table 17. Mean carer's GICS at Week 4, 8, 12, and 16 of Main Phase:
| XEOMIN (6-17 years) N=148 | Placebo (6-17 years) N=72 | |
|---|---|---|
| Week 4* | 0.91 | 0.63 |
| Week 8 | 0.94 | 0.54 |
| Week 12 | 0.87 | 0.47 |
| Week 16 | 0.77 | 0.38 |
* p=0.0320
Efficacy in pediatric patients 2 to 5 years of age is extrapolated from the finding of efficacy in older pediatric patients.
14.2 Upper Limb Spasticity
Upper Limb Spasticity in Adult Patients
The efficacy and safety of XEOMIN for the treatment of upper limb spasticity in adult patients were evaluated in two Phase 3, randomized, multi-center, double-blind studies.
Study 1 and Study 2 were both prospective, double-blind, placebo-controlled, randomized, multi-center trials with an open-label extension period (OLEX) to investigate the efficacy and safety of XEOMIN in the treatment of post-stroke spasticity of the upper limb. For patients who had previously received botulinum toxin treatment in any body region, Study 1 and Study 2 required that ≥12 months and ≥4 months, respectively, had passed since the most recent botulinum toxin administration.
Study 1 consisted of a 12-week main phase followed by three 12-week OLEX treatment cycles for a total exposure duration of 48 weeks. The study included 317 treatment-naïve patients who were at least three months post-stroke in the main study period (210 XEOMIN and 107 placebo). During the main period, XEOMIN (fixed total dose of 400 Units) and placebo were administered intramuscularly to the defined primary target clinical pattern chosen from among the flexed elbow, flexed wrist, or clenched fist patterns and to other affected muscle groups. 296 treated patients completed the main phase and participated in the first OLEX cycle. Each OLEX cycle consisted of a single treatment session (XEOMIN 400 Units total dose, distributed among all affected muscles) followed by a 12-week observation period.
Study 2 consisted of a 12-to-20-week main phase followed by an OLEX period of 48 – 69 weeks, for up to 89 weeks of exposure to XEOMIN. The study included 148 treatment-naïve and pre-treated patients with a confirmed diagnosis of post-stroke spasticity of the upper limb who were at least six months post-stroke (73 XEOMIN and 75 placebo). During the main period, for each patient, the clinical patterns of flexed wrist and clenched fist were treated with fixed doses (90 Units and 80 Units, respectively). Additionally, if other upper limb spasticity patterns were present, the elbow, forearm and thumb muscles could be treated with fixed doses of XEOMIN per muscle. 145 patients completed the main phase and participated in the OLEX period, during which time the dosing of each involved muscle could be adapted individually. During the main and OLEX periods, the maximum total dose per treatment session and 12-week interval was 400 Units.
The average XEOMIN doses injected into specific muscles and the number of injection sites per muscle in Study 1 and Study 2 are presented in Table 18.
Table 18. Doses Administered to Individual Muscles (Main Period) in Adult Upper Limb Spasticity Study 1 and Study 2 Intent to Treat (ITT):
| Muscle Group | Muscle | Study 1 Units Injected | Injection Site Per Muscle | Study 2 Units Injected | Injection Site Per Muscle |
|---|---|---|---|---|---|
| XEOMIN (N=210) Mean±SD | XEOMIN Median (Min; Max) | XEOMIN (N=73) Mean±SD | XEOMIN Median (Min; Max) | ||
| All | Overall | 400 ± 2 Units | -- | 307 ± 77 Units | -- |
| Elbow flexors | Overall | 151 ± 50 Units | 5 (1; 11) | 142 ± 30 Units | 5 (2; 9) |
| Biceps | 90 ± 21 Units | 3 (1; 4) | 80 ± 0 Units | 3 (2; 4) | |
| Brachialis | 52 ± 26 Units | 2 (1; 4) | 50 ± 0 Units | 2 (1; 2) | |
| Brachioradialis | 43 ± 16 Units | 2 (1; 3) | 60 ± 2Units | 2 (1; 3) | |
| Wrist flexors | Overall | 112 ± 43 Units | 4 (1; 6) | 90 ± 0 Units | 4 (4; 4) |
| Flexor carpi radialis | 58 ± 22 Units | 2 (1; 3) | 50 ± 0 Units | 2 (2; 2) | |
| Flexor carpi ulnaris | 56 ± 22 Units | 2 (1; 3) | 40 ± 0 Units | 2 (2; 2) | |
| Finger flexors | Overall | 104 ± 35 Units | 4 (1; 4) | 80 ± 0 Units | 4 (4; 4) |
| Flexor digitorum profundus | 54 ± 19 Units | 2 (1; 2) | 40 ± 0 Units | 2 (2; 2) | |
| Flexor digitorum superficialis | 54 ± 19 Units | 2 (1; 2) | 40 ± 0 Units | 2 (2; 2) | |
| Forearm pronators | Overall | 52 ± 24 Units | 2 (1; 3) | 47 ± 16 Units | 2 (1; 3) |
| Pronator quadratus | 26 ± 13 Units | 1 (1; 1) | 25 ± 0 Units | 1 (1; 1) | |
| Pronator teres | 42 ± 13 Units | 1 (1; 2) | 40 ± 0 Units | 1.5 (1; 2) | |
| Thumb flexors/adductors | Overall | 37 ± 25 Units | 2 (1; 4) | 25 ± 10 Units | 1.5 (1; 3) |
| Adductor pollicis | 14 ± 8 Units | 1 (1; 1) | 10 ± 0 Units | 1 (1; 1) | |
| Flexor pollicis brevis/opponens pollicis | 14 ± 9 Units | 1 (1; 1) | 10 ± 0 Units | 1 (1; 1) | |
| Flexor pollicis longus | 26 ± 16 Units | 1 (1; 2) | 20 ± 0 Units | 1 (1; 1) |
In Study 1, the primary efficacy variable was the change from baseline in Ashworth Scale (AS) score of the primary target clinical pattern determined by the investigator at the Week 4 visit. The Ashworth Scale is a clinical measure of the severity of spasticity by judging resistance to passive movement. The spasticity of the elbow flexors, wrist flexors, finger flexors, and thumb muscles as well as the forearm pronators was assessed on the 0 to 4-point Ashworth scale at each visit. The co-primary efficacy variable of Study 1 was the Investigator's Global Impression of Change Scales (GICS) after 4 Weeks of treatment with XEOMIN or placebo. The GICS is a global measure of a subject's functional improvement. Investigators were asked to evaluate the subject's global change in spasticity of the upper limb due to treatment, compared to the condition before the last injection. The response was assessed using a 7-point Likert scale that ranges from –3 (very much worse) to +3 (very much improved). XEOMIN was considered to be superior to placebo in Study 1 only if statistical significance was reached in both the AS and GICS variables.
The primary efficacy results are displayed in Table 19.
Table 19. Efficacy Results by Patterns of Spasticity in Adult Upper Limb Spasticity Study 1, Week 4:
| Mean Change in Ashworth Scale | ||
|---|---|---|
| XEOMIN (N=171) | Placebo (N=88) | |
| Total Primary Target Clinical Pattern (flexed wrist, flexed elbow, and clenched fist) | -0.9 | -0.5 |
The analysis is based on Last Observation Carried Forward in the Intent To Treat population. p<0.001
A greater percentage of XEOMIN-treated subjects (43%) than placebo-treated subjects (23%) reported 'very much improved' and 'much improved' in their spasticity (see Figure 8).
Figure 8. Investigator's GICS in Adult Upper Limb Spasticity Study 1:

Upper Limb Spasticity in Pediatric Patients
Study 1 was a prospective, double-blind, dose-response, randomized, multi-center trial with an open-label extension period to evaluate the efficacy and safety of XEOMIN for the treatment of upper limb spasticity in pediatric patients. Study 1 enrolled a total of 350 pediatric patients 2 to 17 years of age with upper limb spasticity in one or both upper limbs. In the double-blind main period of Study 1, patients were randomized to one of three dosages of XEOMIN: 2 Units/kg (maximum 50 Units per upper limb), 6 Units/kg (maximum 150 Units per upper limb); or 8 Units/kg (maximum 200 Units per upper limb). The maximum dose, if both upper limbs were treated, respectively was 4 Units/kg (maximum 100 Units), 12 Units/kg (maximum 300 Units), or 16 Units/kg (maximum 400 Units). For treatment of flexed elbow, injection of biceps brachii was mandatory. The investigator could select 1 of the 2 other muscles contributing to spasticity of elbow flexion (i.e., brachialis and brachioradialis) for injection. For patients needing treatment for a flexed wrist, both the flexor carpi radialis and flexor carpi ulnaris were injected. Study 1 used a dose-response design, in which the two highest dosages of XEOMIN (8 Units/kg and 6 Units/kg) were compared to the lowest dosage (2 Units/kg), which served as control. In the absence of a placebo control, the efficacy of the 2 Units/kg dosage of XEOMIN could not be evaluated in Study 1.
The co-primary efficacy variables in Study 1 were the change from baseline on the Ashworth Scale for the primary clinical target pattern (i.e., elbow flexors or wrist flexors), and the Investigator's Global Impression of Change Scale (GICS), both at Week 4. The GICS is a global measure of a subject's functional improvement based on a 7-point Likert scale that ranges from -3 = very much worse to +3 = very much improved.
As displayed in Table 20, the change from baseline in Ashworth Scale score was significantly greater for patients treated with XEOMIN 8 Units/kg than for patients treated with XEOMIN 2 Units/kg. The difference in GICS score between patients treated with XEOMIN 8 Units/kg and those treated with XEOMIN 2 Units/kg did not reach statistical significance. However, the clinical meaningfulness of the difference in Ashworth Scale score change between patients treated with XEOMIN 8 Units/kg and those treated with XEOMIN 2 Units/kg was established by a responder analysis, in which the proportion of patients with a 1-point change or greater on the Ashworth Scale was examined. In that analysis, 86% of patients treated with XEOMIN 8 Units/kg met the responder definition, compared to 71% of patients treated with XEOMIN 2 Units/kg (nominal p value = 0.0099).
There was no significant difference in change from baseline in Ashworth Scale score, GICS score, or proportion of responders between patients treated with XEOMIN 6 Units/kg and those treated with XEOMIN 2 Units/kg. Therefore, the efficacy of a 6 Units/kg dosage of XEOMIN for the treatment of upper limb spasticity in pediatric patients was not established in Study 1.
Table 20. Ashworth Scale and GICS Efficacy Results in Pediatric Upper Limb Spasticity Study 1, Week 4:
| XEOMIN 2 Units/kg (N=87) | XEOMIN 8 Units/kg (N=176) | |
|---|---|---|
| Ashworth Scale | ||
| Mean Change from Baseline at Week 4 | -0.9 | -1.2 |
| LS Mean Difference versus XEOMIN 2 Units/kg (95% CIs) | -- | -0.22* (-0.40, -0.04) |
| GICS | ||
| Mean at Week 4 | 1.6 | 1.7 |
| LS Mean Difference versus XEOMIN 2 Units/kg (95% CIs) | -- | 0.09 (-0.10, 0.28) |
LS = Least Square Mean difference
CI = Confidence Interval
* p-value versus low dose group <0.05
14.3 Cervical Dystonia
XEOMIN has been investigated in a randomized, double-blind, placebo-controlled, multicenter trial in a total of 233 patients with cervical dystonia. Patients had a clinical diagnosis of predominantly rotational cervical dystonia, with baseline Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS) total score ≥20, TWSTRS severity score ≥10, TWSTRS disability score ≥3, and TWSTRS pain score ≥1. For patients who had previously received a botulinum toxin treatment for cervical dystonia, the trial required that ≥10 weeks had passed since the most recent botulinum toxin administration. Patients with swallowing disorders or any significant neuromuscular disease that might interfere with the study were excluded from enrollment. Patients were randomized (1:1:1) to receive a single administration of XEOMIN 240 Units (n=81), XEOMIN 120 Units (n=78), or placebo (n=74). Each patient received a single administration of 4.8 mL of reconstituted study agent (XEOMIN 240 Units, XEOMIN 120 Units, or placebo). The investigator at each site decided which muscles would receive injections of the study agent, the number of injection sites, and the volume at each site. The muscles most frequently injected were the splenius capitis/semispinalis, trapezius, sternocleidomastoid, scalene, and levator scapulae muscles. Table 21 indicates the average XEOMIN dose, and percentage of total dose, injected into specific muscles in the pivotal clinical trial.
Table 21. XEOMIN 120 Units Initial Dose (Units and % of the Total Dose) by Unilateral Muscle Injected During Double Blind Pivotal Phase 3 Study:
| XEOMIN Dose Injected | |||
|---|---|---|---|
| Number of Patients Injected Per Muscle | Median XEOMIN Units | 75th percentile XEOMIN Units | |
| Sternocleidomastoid | 63 | 25 | 35 |
| Splenius capitis/Semispinalis capitis | 78 | 48 | 63 |
| Trapezius | 55 | 25 | 38 |
| Levator scapulae | 49 | 25 | 25 |
| Scalenus (medius and anterior) | 27 | 20 | 25 |
Most patients received a total of 2-10 injections into the selected muscles. Patients were assessed by telephone at one week post-injection, during clinic visits at Weeks 4 and 8, and then by telephone assessments or clinic visits every two weeks up to Week 20.
The mean age of the study patients was 53 years, and 66% of the patients were women. At study baseline, 61% of patients had previously received a botulinum toxin as treatment for cervical dystonia. The study was completed by 94% of study patients. Three patients discontinued the study prematurely due to adverse events: two patients in the 240 Unit group experienced musculoskeletal pain and muscle weakness, and one patient in the 120 Unit group experienced nausea and dizziness.
The primary efficacy endpoint was the change in the TWSTRS total score from baseline to Week 4 post-injection, in the intent-to-treat (ITT) population, with missing values replaced by the patient's baseline value. In the ITT population, the difference between the XEOMIN 240 Unit group and the placebo group in the change of the TWSTRS total score from baseline to Week 4 was -9.0 points, 95% confidence interval (CI) -12.0; -5.9 points; the difference between the XEOMIN 120 Unit group and the placebo group in the change of the TWSTRS total score from baseline to Week 4 was -7.5 points, 95% CI -10.4; -4.6 points.
Figure 9 illustrates the cumulative percentage of patients from each of the three treatment groups who had attained the specified change in TWSTRS Score from baseline versus 4 weeks post-injection. Three change scores have been identified for illustrative purposes, and the percent of patients in each group achieving that result is shown.
Figure 9. Cumulative Percentage of Patients with Specified Changes from Baseline TWSTRS Total Score at Week 4:
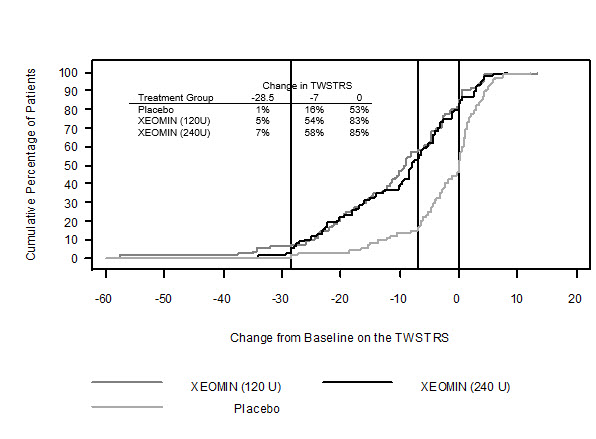
The curves demonstrate that both patients assigned to placebo and XEOMIN have a wide range of responses, but that the active treatment groups are more likely to show greater improvements. A curve for an effective treatment would be shifted to the left of the curve for placebo, while an ineffective or deleterious treatment would be superimposed upon or shifted to the right of the curve for placebo.
Comparison of each XEOMIN group to the placebo group was statistically significant at p<0.001. Initial XEOMIN doses of 120 Units and 240 Units demonstrated no significant difference in effectiveness between the doses. The efficacy of XEOMIN was similar in patients who were botulinum toxin naïve and those who had received botulinum toxin prior to this study.
Examination of age and gender subgroups did not identify differences in response to XEOMIN among these subgroups. There were too few non-white patients enrolled to adequately assess efficacy in other racial populations.
14.4 Blepharospasm
Treatment-Naïve Patients
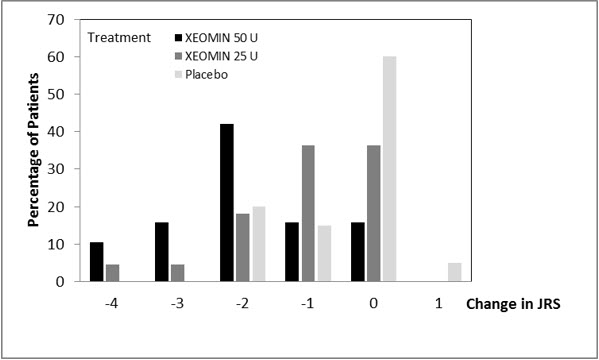
The efficacy and safety of XEOMIN for the treatment of blepharospasm in treatment-naïve patients were evaluated in Study 1, a randomized, double-blind, placebo-controlled, multi-center trial in a total of 61 patients. Patients had a clinical diagnosis of blepharospasm, with a baseline Jankovic Rating Scale (JRS) severity subscore ≥2. Patients were defined as treatment-naïve if at least 12 months had passed since their last botulinum toxin treatment for blepharospasm. During the placebo-controlled phase, a fixed total dose of 25 Units XEOMIN (n=22), 50 Units XEOMIN (n=19), or placebo (n=20) was administered intramuscularly at 6 injection sites per eye (Figure 6). Of the 61 patients randomized, 55 patients completed the placebo-controlled phase. Patients only continued to the open-label extension (OLEX) period if they had a confirmed need for a re-injection by week 20 of the placebo-controlled phase. A total of 39 patients entered and completed the OLEX phase.
The primary efficacy variable was the change from baseline in JRS Severity subscore determined at Week 6 after the injection. The 50 Unit treatment group demonstrated statistically significant improvements compared to placebo, with a difference of -1.2 (p=0.0004). The change from baseline in the JRS Severity subscore for the 25 Unit treatment group 6 weeks after the injection was not statistically significant, with a difference of -0.5 (p=0.1452) compared to placebo (see Figure 10).
Figure 10. Frequency Distribution of Changes from Baseline JRS Severity Subscore at Week 6 for Treatment-Naïve Patients:
Pre-Treated Patients
The efficacy and safety of XEOMIN for the treatment of blepharospasm patients pre-treated with onabotulinumtoxinA (Botox) were evaluated in Study 2, a randomized, double-blind, placebo-controlled, multi-center trial in a total of 109 patients. Patients had a clinical diagnosis of benign essential blepharospasm, with baseline JRS Severity subscore ≥2, and a stable satisfactory therapeutic response to previous administrations of onabotulinumtoxinA (Botox). At least 10 weeks had to have elapsed since the most recent onabotulinumtoxinA administration. Patients with any significant neuromuscular disease that might interfere with the study were excluded from enrollment. Patients were randomized (2:1) to receive a single administration of XEOMIN (n=75) or placebo (n=34). Each patient in the XEOMIN group received a XEOMIN treatment (dose, volume, dilution, and injection sites per muscle) that was similar to the most recent onabotulinumtoxinA injection sessions prior to study entry. The highest dose permitted in this study was 100 Units (50 Units per eye); the mean XEOMIN dose was 33 Units per eye.
In Table 22 the most frequently injected sites, the median dose per injection site, and the median number (and range) of injection sites per eye are presented.
Table 22. Median Dose and Median Number of Injection Sites per Eye (Blepharospasm):
| Injection Area | Median Units XEOMIN | Median Number of Injection Sites (Min-Max) |
|---|---|---|
| Temporal Area | 13 | 2 (1–6) |
| Eyebrow Area | 5 | 1 (1–4) |
| Upper Lid Area | 10 | 2 (1–4) |
| Lower Lid Area | 8 | 2 (1–3) |
| Orbital Rim | 5 | 1 (1–3) |
Patients were assessed during clinic visits at Weeks 3 and 6, and then by telephone or at clinic visits every two weeks up to Week 20.
The mean age of the study patients was 62 years, and 65% of the patients were women. The study was completed by 94% of study patients. Approximately one third of patients had other dystonic phenomena; in all but 1% this was limited to facial, cervical, perioral and mandibular muscles. No patients discontinued the study prematurely due to adverse events.
The primary efficacy endpoint was the change in the JRS Severity subscore from baseline to Week 6 post-injection, in the intent-to-treat (ITT) population, with missing values replaced by the patient's most recent value (i.e., last observation carried forward). In the ITT population, the difference between the XEOMIN group and the placebo group in the change of the JRS Severity subscore from baseline to Week 6 was -1.0 (95% CI -1.4; -0.5) points. Comparison of the XEOMIN group to the placebo group was statistically significant at p<0.001.
Figure 11. Frequency Distribution of Changes from Baseline JRS Severity Subscore at Week 6:
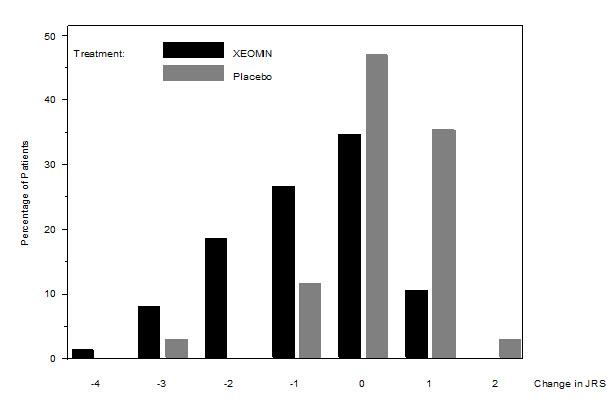
Examination of age and gender subgroups did not identify substantial differences in response to XEOMIN among these subgroups. There were too few non-white patients enrolled to adequately assess efficacy in other racial populations.
14.5 Glabellar Lines
Two identically designed randomized, double-blind, multi-center, placebo controlled clinical trials (Studies GL-1 and GL-2) were conducted to evaluate XEOMIN for use in the temporary improvement of moderate to severe glabellar lines. The studies enrolled 547 healthy patients (≥18 years old) with glabellar lines of at least moderate severity at maximum frown. Three hundred sixty six subjects were treated with 20 Units of XEOMIN and 181 subjects were treated with placebo. Subjects were excluded if they had marked ptosis, deep dermal scarring, or an inability to lessen glabellar lines, even by physically spreading them apart. The mean age of study subjects was 46 years. The majority of patients were female (86% and 93% in Studies GL-1 and GL-2, respectively), and predominantly Caucasian (89% and 65% respectively). The study subjects received either 20 Units of XEOMIN or an equal amount of placebo. The total dose was delivered in 5 equally divided intramuscular injections of 4 Units each to specific sites (see Figure 7). Subjects were followed up for 120 days.
Investigators and subjects assessed efficacy at maximum frown on Day 30 of treatment using a 4-point scale (0=none, 1=mild, 2=moderate, 3=severe). Composite treatment success was defined as a 2-grade improvement on this scale compared to baseline for both the investigator's and subject's assessments on Day 30. The percentage of subjects with treatment success was greater on the XEOMIN arm than the placebo arm at Day 30 in both studies (see Table 23). The percentage of subjects with composite treatment success at each visit is presented in Figure 12.
Table 23. Treatment Success at Day 30 (at Least 2 Grades Improvement from Baseline at Maximum Frown):
| GL-1 | GL-2 | |||
|---|---|---|---|---|
| XEOMIN (N=184) | Placebo (N=92) | XEOMIN (N=182) | Placebo (N=89) | |
| Composite Treatment Success* | 111 (60%) | 0 (0%) | 87 (48%) | 0 (0%) |
| Investigator Assessment | 141 (77%) | 0 (0%) | 129 (71%) | 0 (0%) |
| Subject Assessment | 120 (65%) | 0 (0%) | 101 (55%) | 1 (1%) |
* Success on both the Investigator and Subject Assessments
Figure 12. Percentage of Subjects with Composite Treatment Success by Visit – Observed Cases (GL-1 and GL-2):

© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.